Moulay El Hassane EL MOUKRIE. Ingénieur procédés industriels et génie chimique à la recherche d'opportunités.
Site personnel de Mr Moulayelhassane EL MOUKRIE, Ingénieur des Procédés Industriels et Génie Chimique. A la recherche d'opportunités professionnelles. Vous y trouvez son profil: Formation, Expériences, Compétences, Connaissances et ses Centres d'Intérêt.
Gerald Steiner, Bernhard Geissler, and Nils Haneklaus
Introduction
Les États-Unis sont le plus grand consommateur mondial d’uranium. En 2017, la très grande majorité, soit 93 %, de cet uranium était importée. Une enquête récente menée par l’administration actuelle a conclu que les importations étrangères d’uranium et l’approvisionnement en produits connexes — essentiels pour l’arsenal nucléaire américain, la marine hauturière et les centrales électriques — ne constituaient pas une menace pour la sécurité nationale. Toutefois, le déclin de l’exploitation minière nationale de l’uranium est considéré comme une préoccupation importante.
Les exploitants miniers d’uranium aux États-Unis ont lancé cette enquête dans l’espoir d’obtenir des quotas sur les importations étrangères d’uranium, ce qui leur permettrait de mieux concurrencer les entreprises étrangères, souvent publiques et fortement subventionnées. Des quotas, s’ils étaient mis en place, pourraient en effet relancer l’exploitation minière de l’uranium aux États-Unis. Ils pourraient également redonner un intérêt à la récupération de l’uranium à partir de ressources non conventionnelles, notamment les phosphates, qui, à leur apogée, ont contribué à près de 20 % des besoins du pays en uranium dans les années 1980. Cette voie pourrait redevenir rentable et être mise en œuvre beaucoup plus rapidement que l’ouverture de nouvelles mines d’uranium, lesquelles finiraient nécessairement par devoir être implantées quelque part, avec les oppositions locales que cela implique.
Les phosphates — et plus précisément ici les roches phosphatées sédimentaires — peuvent contenir des quantités considérables d’uranium associé, aussi bien en termes de concentrations que de quantités globales. La roche phosphatée est le quatrième matériau le plus extrait au monde et elle est principalement utilisée, à plus de 90 % à l’échelle mondiale, pour la production d’engrais minéraux.
Les techniques permettant de récupérer l’uranium à partir de l’acide phosphorique, produit intermédiaire liquide de la fabrication des engrais phosphatés, sont bien connues et ont été utilisées aux États-Unis, ainsi que, dans une moindre mesure, ailleurs, à l’échelle industrielle jusqu’à la fin des années 1990, lorsque les prix de l’uranium se sont effondrés, rendant la récupération non rentable pour les producteurs d’engrais.
Quantités d’uranium dans les phosphates
La sensibilisation accrue aux enjeux environnementaux, la sécurité énergétique nationale et la possible hausse des prix de l’uranium ont suscité un regain d’intérêt mondial pour cette technologie. Gabriel et al. ainsi qu’Ulrich et al. ont estimé que les producteurs d’engrais phosphatés pourraient fournir un peu plus de 15 % des besoins mondiaux en uranium en temps de paix. Des études similaires ont été réalisées pour l’Argentine, où l’uranium récupéré comme coproduit de la production d’engrais phosphatés pourrait couvrir 8 à 9 % des besoins en uranium ; pour l’Union européenne, environ 2 % des besoins en uranium ; et pour les États-Unis, environ 10 % des besoins en uranium.
Dans ces études, les quantités d’uranium récupérables à partir des phosphates dépasseraient souvent la production actuelle issue des mines d’uranium conventionnelles nationales. Kim et al. ont par exemple estimé que 5,5 millions de livres de U₃O₈, soit davantage que la production nationale américaine de 2014, qui était de 4,9 millions de livres de U₃O₈, auraient pu être fournies par l’industrie américaine des phosphates.
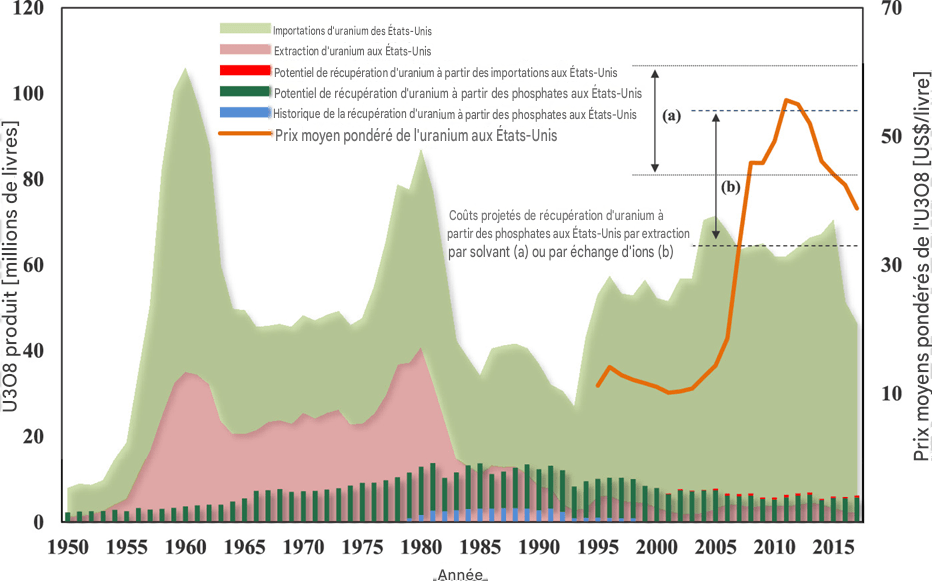
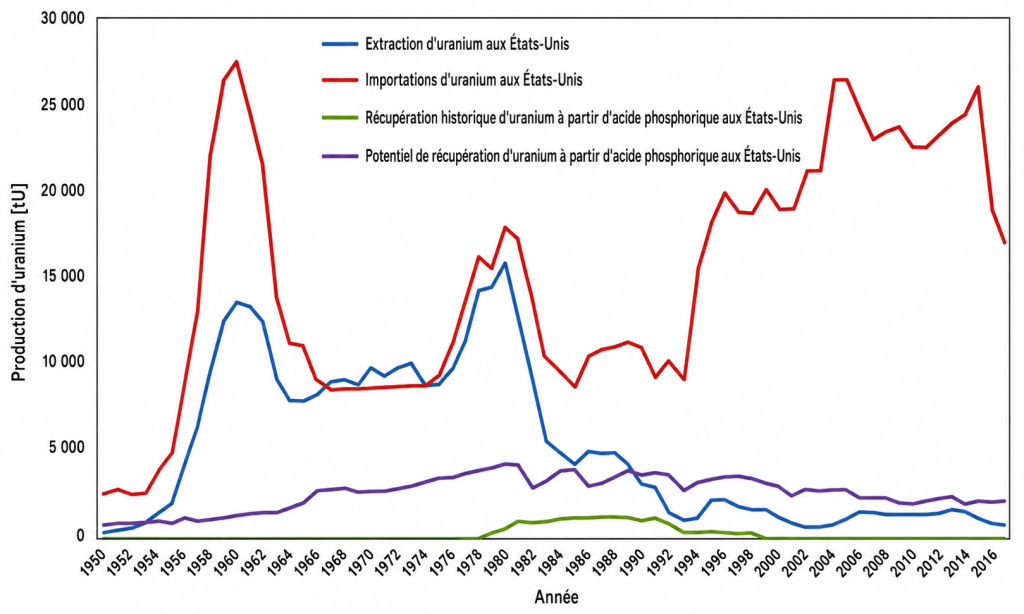
La figure 1 compare la production annuelle d’uranium aux importations annuelles d’uranium aux États-Unis. Elle indique également la récupération historique de l’uranium à partir des phosphates ainsi que le potentiel de récupération de l’uranium à partir des phosphates aux États-Unis. Avec la diminution de l’exploitation minière de l’uranium en 1990, les quantités d’uranium qui auraient pu être récupérées à partir des phosphates étaient supérieures aux quantités effectivement extraites des mines.
Dans la figure 1, sont pris en compte l’uranium provenant de toutes les mines de phosphate des États-Unis ainsi que l’uranium contenu dans les importations de roches phosphatées. Bien que sa concentration soit relativement élevée en Floride, environ 160 mg/kg — à comparer à la mine de Rössing en Namibie, l’une des plus anciennes exploitations commerciales d’uranium en activité et la cinquième plus grande mine d’uranium commerciale, avec des concentrations moyennes d’uranium de 200 à 300 mg/kg et une teneur moyenne de 0,003 mg/kg d’uranium dans l’eau de mer — cette teneur justifierait une récupération. Toutefois, la concentration peut être moins élevée dans d’autres régions, par exemple 107 mg/kg dans l’Idaho et 65 mg/kg en Caroline du Nord. Dans ces conditions, une récupération rentable de l’uranium serait plus difficile.
Dans ce contexte, il convient de noter que la majorité de l’uranium radiotoxique, soit 70 à 80 %, qui n’est pas récupéré est — et continuera d’être — dispersée avec les engrais sur les sols agricoles. Le reste se retrouve dans le flux de déchets de phosphogypse, c’est-à-dire une boue composée de sulfate de calcium et d’eau, présentant de faibles niveaux de radioactivité et donc des possibilités d’utilisation limitées.
Figure 1. Importations américaines d’uranium, production nationale d’uranium, récupération potentielle et historique de l’uranium à partir des phosphates, prix moyens pondérés de l’uranium, ainsi que coûts minimaux et maximaux projetés pour la récupération de l’uranium par extraction par solvant (a) et par échange d’ions (b) aux États-Unis. Source : EIA (8) et Beltrami et al. (1).
Économie de la récupération
Malgré des prix de l’uranium relativement faibles, les propriétaires et exploitants de réacteurs électronucléaires civils aux États-Unis ont acheté, en 2018, un total de 40 millions de livres d’équivalent U₃O₈ auprès de fournisseurs américains et étrangers, à un prix moyen pondéré de 38,81 dollars américains par livre de U₃O₈. Près de 42 % de cet uranium provenait du Canada et d’Australie ; environ 40 % provenait du Kazakhstan, de Russie et d’Ouzbékistan ; et près de 10 % provenait des États-Unis. L’uranium d’origine américaine a été livré à un prix moyen pondéré de 45,26 dollars américains par livre de U₃O₈.
Dans ce contexte, les coûts de récupération de l’uranium à partir de l’acide phosphorique, au moyen d’une technologie d’extraction par solvants industriellement éprouvée, sont estimés aujourd’hui aux États-Unis entre 44 et 61 dollars américains par livre de U₃O₈. Ces coûts se situent donc à la limite de la rentabilité économique.
La récupération de l’uranium au moyen de procédés fondés sur l’échange d’ions n’est pas encore une technologie commerciale éprouvée pour la récupération de l’uranium, mais elle est actuellement testée à l’échelle pilote aux États-Unis par PhosEnergy, une société australienne. Elle pourrait réduire davantage les coûts, jusqu’à environ 33 à 54 dollars américains par livre de U₃O₈.
Des économies encore plus importantes pourraient être réalisées si l’uranium radiotoxique était lixivié directement, avant l’étape d’attaque/digestion, à partir de la roche phosphatée enrichie. Cela permettrait de rendre non seulement l’engrais final, mais aussi le sous-produit phosphogypse, pratiquement exempts d’uranium.
Bien que le gypse soit un matériau de construction largement utilisé, le phosphogypse est généralement stocké indéfiniment en raison de sa faible radioactivité, qui résulte de la présence naturelle d’uranium et, dans une moindre mesure, de thorium dans les roches phosphatées traitées. Quelque 100 à 300 millions de tonnes de phosphogypse sont produites chaque année dans le monde. Rendre ce matériau disponible pour des utilisations non contestables pourrait donc permettre à l’industrie des engrais d’économiser des centaines de millions de dollars américains en coûts annuels de stockage qui s’accumulent autrement.
Par ailleurs, au regard des coûts directs précédemment mentionnés pour la récupération de l’uranium à partir des phosphates, qui peuvent être estimés aujourd’hui, il convient de souligner que nous pourrions être confrontés à des coûts indirects croissants liés à la purification des eaux souterraines contaminées par l’uranium issu des engrais si les pratiques actuelles restent inchangées.
Quo vadis, récupération de l’uranium ?
La récupération de l’uranium à partir des phosphates est déjà une bonne idée du point de vue de la conservation des ressources et des Objectifs de développement durable, ODD, des Nations unies. Le cadre établi par les décideurs politiques américains déterminera si l’uranium sera ou non à nouveau récupéré au cours de la production d’engrais.
Dans ce contexte, des quotas sur les importations étrangères d’uranium aux États-Unis pourraient, de manière consciente ou non, fournir un cadre incitatif à la récupération de l’uranium pendant la production américaine d’engrais, et ainsi rendre à nouveau attractive la récupération de l’uranium à partir des phosphates.
Informations sur les auteurs
Auteur correspondant
Nils Haneklaus — Danube University Krems, Krems, Autriche, et RWTH Aachen University, Aix-la-Chapelle, Allemagne ; ORCID : orcid.org/0000-0002-673-0376 ; Email : nils.haneklaus@rwth-aachen.de
Autres auteurs
Gerald Steiner — Danube University Krems, Krems, Autriche
Bernhard Geissler — Danube University Krems, Krems, Autriche
Les auteurs déclarent ne pas avoir d’intérêts financiers concurrents.
Références
(1) Beltrami, D.; Cote, G.; Mokhtari, H.; Courtaud, B.; Moyer, B. A.; Chagnes, A. Recovery of Uranium from Wet Process Phosphoric Acid by Solvent Extraction. Chem. Rev.2014, 114, 12002–12023.
(2) Singh, D. K.; Mondal, S.; Chakravartty, J. K. Recovery of Uranium From Phosphoric Acid: A Review. Solvent Extr. Ion Exch.2016, 34, 201–25.
(3) Gabriel, S.; Baschwitz, A.; Mathonnière, G.; Fizaine, F.; Eleouet, T. Building future nuclear power fleets: The available uranium resources constraint. Resour. Policy2013, 38, 458–469.
(4) Ulrich, E.; Schnug, H.-M.; Prasser, E. Frossard Uranium endowments in phosphate rock. Sci. Total Environ.2014, 478, 226–234.
(5) López, L.; Castro, L. N.; Scasso, R. A.; Grancea, L.; Tulsidas, H.; Haneklaus, N.; Nacional, C.; Atamica, D. E.; Libertador, A.; Aires, C. B. Uranium supply potential from phosphate rocks for Argentina’s nuclear power fleet. Resour. Policy2019, 62, 397–404.
(6) Tulsidas, H.; Gabriel, S.; Kiegiel, K.; Haneklaus, N. Uranium resources in EU phosphate rock imports. Resour. Policy2019, 61, 151–156.
(7) Kim, H.; Eggert, R. G.; Carlsen, B. W.; Dixon, B. W. Potential uranium supply from phosphoric acid: A U.S. analysis comparing solvent extraction and ion exchange recovery. Resour. Policy2016, 49, 222–231.
(10) Al Khaledi, N.; Taha, M.; Hussein, A.; Hussein, E.; El Yahyaoui, A.; Haneklaus, N. Direct leaching of rare earth elements and uranium from phosphate rocks. IOP Conf. Ser.: Mater. Sci. Eng.2019, 479, 012065.
Production d’acide phosphorique à l’aide d’acides autres que l’acide sulfurique
Production d’acide phosphorique par procédé au four électrique
Production d’acide phosphorique par procédé au haut-fourneau
Utilisation du gypse coproduit
Dans la production d’acide phosphorique par voie humide, environ 5 tonnes, sur base sèche, de phosphogypse coproduit sont générées par tonne de P₂O₅ récupéré sous forme d’acide phosphorique. Seuls environ 15 % de ce phosphogypse sont réutilisés. Le phosphogypse retient au moins 20 % d’eau libre en masse. Les principaux modes de valorisation du phosphogypse sont les suivants :
○ Produire du sulfate d’ammonium par réaction du gypse avec l’ammoniac et le dioxyde de carbone.
○ Produire du ciment et de l’acide sulfurique par calcination du gypse avec du coke et de l’argile ou du schiste.
○ Produire du plâtre ou des plaques de plâtre destinés aux matériaux de construction, ou fabriquer des blocs pressés ou moulés pour des usages de construction.
○ L’utiliser dans le ciment comme retardateur de prise.
○ L’utiliser comme charge pour engrais.
○ L’utiliser en application directe sur les terres agricoles lorsque le sol l’exige.
Valorisation du fluor
La plupart des roches phosphatées de type fluoroapatite contiennent une quantité significative de fluor, généralement 3 à 4 % de F en masse. Dans certains cas, jusqu’à 60 % de ce fluor peut être dégagé au cours de la fabrication de l’acide phosphorique par voie humide. Le reste du fluor est retenu dans le gypse, principalement en fonction de la composition de la roche, et la majeure partie de la fraction restante se retrouve dans l’acide filtré [30].
Le fluor est libéré à différentes étapes des procédés de fabrication de l’acide phosphorique : à partir de la surface de la bouillie réactionnelle dans le réacteur, dans le refroidisseur flash, ainsi que dans l’atelier de concentration.
Dans un procédé dihydrate, les proportions sont les suivantes :
○ Surface de la bouillie : 3 à 5 %
○ Refroidisseur flash : 8 à 10 %
○ Atelier de concentration : 15 à 20 % jusqu’à 45 % de P₂O₅
Dans les procédés hémihydrate ou hémidihydrate, produisant un acide à 45 % de P₂O₅ au niveau du filtre, les proportions sont les suivantes :
○ Surface de la bouillie : 12 à 15 %
○ Refroidisseur flash : 15 à 20 %
Dans un cas typique, environ 50 kg de F sont volatilisés par tonne de P₂O₅, généralement sous forme de SiF₄ ou de HF, ou d’un mélange des deux. Les compositions des vapeurs émises par la section réaction/filtration et par l’évaporateur sont différentes.
Lors de l’attaque acide de la roche phosphatée, le fluorure est d’abord converti en fluorure d’hydrogène, lequel réagit ensuite avec la silice active pour former du tétrafluorure de silicium :
4HF + SiO₂ = SiF₄ + 2H₂O
Celui-ci est partiellement émis sous forme de vapeur préférentiellement au fluorure d’hydrogène, en raison de sa pression de vapeur plus élevée. Le reste réagit davantage pour former de l’acide fluosilicique, qui demeure dans l’acide produit, ou des fluosilicates insolubles, lesquels sont éliminés dans le gâteau de filtration.
3SiF₄ + 2H₂O = 2H₂SiF₆ + SiO₂
Si la vapeur de tétrafluorure de silicium quittant la section de réaction est lavée à l’eau, cette réaction se produit également dans le laveur, entraînant la formation de silice insoluble. Cette silice provoque des dépôts et peut finir par colmater le laveur.
Par conséquent, la procédure habituelle consiste à effectuer le lavage avec une solution diluée d’acide fluosilicique, dans laquelle la silice reste en suspension colloïdale. Un laveur de conception adaptée, tel qu’un Venturi cyclone ou une tour vide, pratiquement dépourvu d’obstructions internes susceptibles de s’encrasser, doit être utilisé au premier étage afin d’éliminer la majeure partie du fluor.
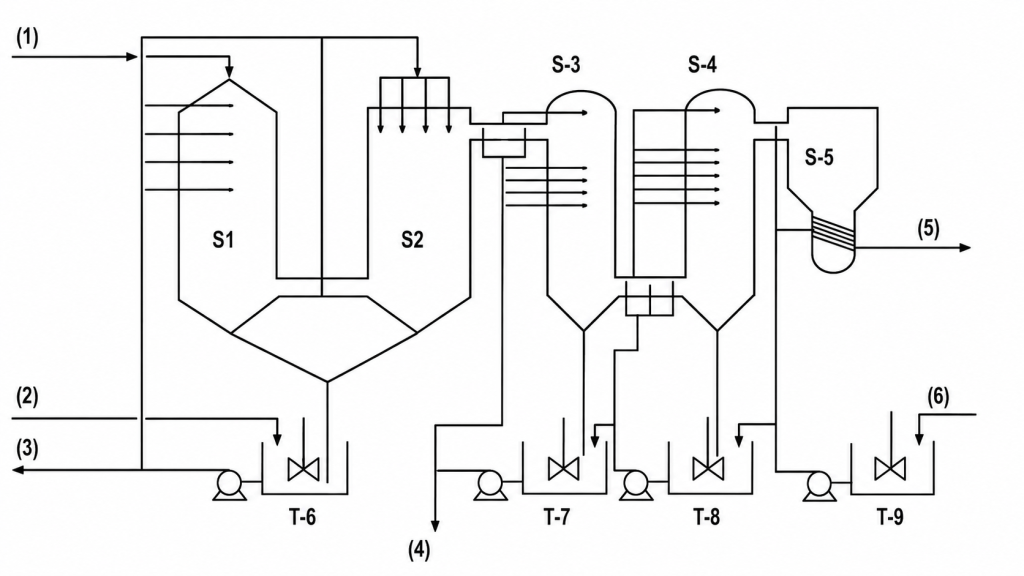
Un second étage, très efficace, tel qu’une tour de pulvérisation ou une tour équipée d’un garnissage mobile, élimine la majeure partie des traces résiduelles de fluor et évite l’encrassement (Figure 1). Les vapeurs plus diluées, telles que l’air provenant de la hotte au-dessus du premier étage de filtration et du premier séparateur de filtrat, peuvent être introduites directement dans le laveur du second étage.
Figure 1. Système de lavage du fluor en deux étages sous licence Norsk Hydro
(1) – GAZ PROVENANT DU REFROIDISSEUR FLASH (2) – APPOINT D’ACIDE FLUOSILICIQUE (3) – ACIDE FLUOSILICIQUE CONTAMINÉ VERS LE RÉACTEUR (4) – VERS LE RÉSERVOIR DE STOCKAGE DU PREMIER ÉTAGE (5) – GAZ C/T VERS LE CONDENSEUR DU REFROIDISSEUR FLASH (6) – LIQUEUR PROVENANT DU REFROIDISSEUR DE L’ÉTAGE FINAL
Légende :
S-1 Laveur Venturi S-2 Séparateur de gouttelettes S-3 Laveur du premier étage S-4 Laveur du second étage S-5 Séparateur de gouttelettes T-6 – T-9 Cuves de garde
Environ deux tiers du fluor contenu dans l’acide filtré sont volatilisés lorsque l’acide est concentré jusqu’à la qualité commerciale, soit 50 à 55 % de P₂O₅. Dans les conditions de haute température et de pression réduite rencontrées dans les évaporateurs sous vide, l’acide fluosilicique se décompose, et du tétrafluorure de silicium ainsi que du fluorure d’hydrogène sont dégagés.
H₂SiF₆ = 2HF + SiF₄
Toutefois, lorsque les concentrations d’acide sont inférieures à 50 % de P₂O₅, davantage de tétrafluorure de silicium est dégagé, formant de la silice dans l’acide fluosilicique en circulation, en raison de la relation entre sa pression de vapeur et celle du fluorure d’hydrogène.
À des concentrations d’acide de 50 à 55 % de P₂O₅, le rapport molaire HF:SiF₄ est approximativement égal à 2, tandis qu’à des concentrations d’acide plus élevées, le fluorure d’hydrogène prédomine [30].
Le risque de problèmes liés aux dépôts de silice dans le laveur est minimal lorsque le rapport HF:SiF₄ est au moins égal à 2, car le seul produit du lavage sera alors l’acide fluosilicique. En revanche, il n’est pas souhaitable de laisser ce rapport dépasser excessivement 2, car le produit contiendrait alors du fluorure d’hydrogène libre.
Comme la solution de lavage utilisée est de l’acide fluosilicique, il est souhaitable de maintenir une concentration élevée, notamment du point de vue des coûts de transport et de l’utilisation du produit. Afin d’éviter la dilution par l’eau provenant de l’évaporateur, le laveur, intercalé entre l’évaporateur et le condenseur, doit fonctionner à une température élevée à laquelle l’eau ne se condense pas de manière significative.
En pratique, cela fixe une limite maximale d’environ 18 à 22 % de H₂SiF₆ pour la concentration du milieu de lavage, car à des concentrations plus élevées, les pressions de vapeur à l’équilibre du fluorure d’hydrogène et du tétrafluorure de silicium deviennent trop importantes pour permettre une élimination efficace du fluor.
Le type de laveur à tour vide pulvérisée, initialement développé par Swift and Co., puis perfectionné par Fisons et d’autres sociétés, est souvent utilisé. Le laveur est généralement monté au-dessus de l’évaporateur et reçoit les vapeurs issues du séparateur d’entraînement, lequel renvoie vers le circuit acide les gouttelettes ou brouillards d’acide phosphorique.
Les laveurs de fluor fonctionnent généralement avec une efficacité de 90 à 93 %. Le reste des vapeurs fluorées est en grande partie éliminé dans le condenseur traversé ensuite par les vapeurs. Le condenseur est généralement du type barométrique à contact direct ; toutefois, après refroidissement, l’eau doit être dirigée vers une installation de traitement des eaux usées afin d’éliminer la contamination.
De nombreux procédés de récupération de composés fluorés commercialisables ont été proposés et développés expérimentalement ; certains sont utilisés à l’échelle industrielle. La description complète de ces procédés dépasse le cadre de cette ressource.
Le fluor est généralement récupéré sous forme d’une solution aqueuse d’acide fluosilicique, H₂SiF₆. Sa concentration peut atteindre 20 à 25 %. Dans certains pays, l’acide fluosilicique est utilisé directement pour la fluoration des réseaux municipaux d’eau potable afin de prévenir la carie dentaire. L’acide est expédié vers diverses municipalités dans des wagons-citernes revêtus de caoutchouc.
Les sels de l’acide fluosilicique, tels que les fluosilicates de sodium, de potassium et d’ammonium, ont diverses applications et peuvent être facilement produits à partir de cet acide. Le fluosilicate de sodium est utilisé pour la fluoration des réseaux municipaux d’eau potable, mais l’acide fluosilicique est généralement préféré.
Des procédés ont été développés pour produire du fluorure d’aluminium AlF₃ et de la cryolithe Na₃AlF₆ à partir d’acide fluosilicique, produits utilisés en quantités importantes par l’industrie de l’aluminium. Toutefois, ces matériaux doivent être très purs pour cet usage ; en particulier, les teneurs en silicium et en phosphore doivent être très faibles. Cette exigence complique la production à partir des coproduits de l’industrie phosphatière.
Le fluorure de calcium peut être produit et utilisé à la place du spath fluor naturel pour la production de fluorure d’hydrogène HF, qui constitue la matière première de base pour la fabrication de nombreux composés fluorés organiques et inorganiques, ou pour des applications métallurgiques [31]. Il existe également des procédés permettant de produire des solutions d’acide fluorhydrique ou du fluorure d’hydrogène anhydre à partir de solutions d’acide fluosilicique.
En général, l’économie de production de composés fluorés de qualité chimique à partir d’acide fluosilicique coproduit est marginale à défavorable pour les petites unités d’acide phosphorique. Toutefois, les grandes unités peuvent rendre cette récupération rentable. Lorsque plusieurs usines d’acide phosphorique fonctionnent dans un rayon d’expédition économiquement acceptable, l’acide fluosilicique brut peut être expédié vers un point central pour la production de composés fluorés raffinés. Pour plus de détails sur l’utilisation du fluor, voir la référence [30].
Purification de l’acide phosphorique
Pour la plupart des procédés de production d’engrais, la purification de l’acide phosphorique par voie humide n’est pas nécessaire. Cependant, deux utilisations courantes dans les engrais peuvent nécessiter une purification partielle :
L’acide de qualité marchande, expédié par rail, barge ou navire océanique, et souvent stocké dans des terminaux d’expédition et de réception, doit être suffisamment purifié afin de minimiser la formation de précipités insolubles, ou boues, pendant le transport et le stockage.
L’acide phosphorique destiné à la production d’engrais liquides, tels que les solutions de polyphosphate d’ammonium, nécessite parfois une purification partielle afin d’éviter la formation de précipités lors de l’ammoniation ou pendant le stockage de la solution ammoniée.
Bien que le polyphosphate d’ammonium séquestre la plupart des impuretés courantes, des quantités excessives de certaines impuretés, notamment le magnésium et la matière organique, provoquent la formation de précipités. Les acides superphosphoriques ne forment généralement pas de boues, mais le magnésium et le titane sont connus pour pouvoir provoquer des précipités générateurs de boues.
Une fraction importante des boues présentes dans la plupart des acides marchands générant des boues est constituée du composé :
(Fe,Al)₃KH₁₄(PO₄)₈ · 4H₂O
Il précipite lentement sur une période de plusieurs semaines ; par conséquent, de longues durées de stockage sont nécessaires pour assurer un achèvement raisonnable de la réaction de précipitation.
Dans la production de tripolyphosphate de sodium et d’autres phosphates condensés destinés à être utilisés comme adjuvants dans les détergents, la blancheur du produit et l’ensemble des ions métalliques, à l’exception du sodium et du potassium, constituent les principales préoccupations.
Pour les usages en transformation alimentaire humaine et dans la production de compléments pour l’alimentation animale, la teneur en impuretés toxiques doit être réduite à des niveaux insignifiants. Ces impuretés comprennent le fluor, l’arsenic et les métaux lourds éventuellement présents dans la roche phosphatée et l’acide sulfurique.
Lorsque l’acide purifié n’est requis que pour la production de phosphates, l’élimination des impuretés est facilitée par neutralisation, car de nombreuses impuretés précipitent rapidement sous forme de phosphates insolubles.
Lorsqu’il n’est pas possible de neutraliser l’acide, il est nécessaire de recourir à un traitement physique tel que l’extraction par solvant ou l’échange d’ions, avec ou sans traitement chimique. Les fabricants d’acide phosphorique purifiant l’acide phosphorique pour la vente se sont orientés vers ce type de traitement, généralement basé sur l’extraction par solvant.
Dans la plupart des cas où des réactifs chimiques sont utilisés, par exemple pour l’élimination de l’arsenic, ils réagissent uniquement avec l’impureté concernée et ne modifient pas chimiquement l’acide phosphorique.
Élimination des matières organiques
Dans l’environnement oxydant et déshydratant de la section d’attaque, les impuretés organiques tendent à se décomposer en carbone ; si la roche présente une teneur organique relativement élevée, l’acide obtenu sera noir.
Deux méthodes permettant d’éliminer cette coloration comprennent la calcination de la roche, qui détruit une bonne partie de la matière organique, et le traitement de l’acide au charbon actif, avec ou sans agent floculant.
La réalisation de ce dernier traitement à température modérée, soit 60 à 80 °C, augmente l’efficacité de l’élimination des matières organiques au point qu’il peut être possible d’éviter la calcination de certaines roches à forte teneur organique.
D’autres techniques proposées reposent sur l’utilisation d’un agent floculant spécial ou de peroxyde d’hydrogène en présence d’un catalyseur constitué d’oxydes métalliques mixtes.
Défluoration
Même lorsque l’acide phosphorique par voie humide est concentré jusqu’au stade d’acide superphosphorique, il contient encore trop de fluor pour la production de phosphates destinés à l’alimentation animale, bien que les niveaux des autres impuretés puissent être acceptables pour l’alimentation animale.
Pour la préparation d’aliments pour animaux, il est souhaitable que le rapport phosphore/fluor soit d’au moins :
100 P : 1 F
Or l’acide phosphorique normal par voie humide présente un rapport P:F compris entre 15:1 et 54:1, selon sa concentration, la composition de la roche à partir de laquelle il est produit et ses conditions de production.
Plusieurs procédures ont été proposées pour éliminer le fluor de l’acide phosphorique, fondées sur la volatilisation ou la précipitation chimique, dans ce dernier cas avec ou sans élimination d’autres impuretés.
Procédés par solvants
Précipitation
Dans ce type de procédé, une grande quantité de solvant organique miscible à l’eau est ajoutée à l’acide phosphorique, ce qui provoque la décantation de nombreuses impuretés dissoutes hors de la solution et permet la séparation du mélange acide-solvant.
En particulier, si du méthanol est utilisé, des ions de métaux alcalins ou d’ammonium doivent être présents en quantités suffisantes pour convertir les impuretés boueuses en sels doubles moins solubles. D’autres solvants pouvant être utilisés comprennent l’acétone, la méthyléthylcétone, d’autres alcools inférieurs et le dioxane.
L’inconvénient de ce type de procédé est que l’acide phosphorique doit être séparé d’un grand volume de solvant, généralement par stripping, ce qui est coûteux.
Extraction
La méthode alternative de purification de l’acide phosphorique au moyen de solvants organiques est l’extraction liquide-liquide. L’acide phosphorique brut est mis en contact à contre-courant avec un solvant partiellement ou substantiellement non miscible à l’eau dans une série de mélangeurs-décanteurs ou de colonnes.
En général, le coefficient de partage de l’acide phosphorique entre la solution aqueuse et le solvant est très défavorable ; ainsi, l’acide brut est généralement concentré avant traitement, et seule une proportion relativement faible de l’acide est extraite.
Le raffinat partiellement appauvri restant, contenant la majorité des impuretés, est habituellement utilisé dans la fabrication d’engrais.
Selon le degré de purification requis, l’extrait peut être lavé avec de l’acide phosphorique purifié, puis il est généralement mis en contact avec de l’eau, dans laquelle passe la majeure partie de l’acide.
Le produit final est un acide légèrement plus faible, mais néanmoins plus pur que l’acide brut initial, ainsi qu’une phase solvant appauvrie, qui est recyclée.
Lorsqu’il n’existe aucune possibilité d’utiliser le raffinat dans la production d’engrais, celui-ci doit être fortement acidifié avec de l’acide sulfurique avant une extraction supplémentaire ; la majeure partie du P₂O₅ est alors transférée dans la phase solvant. Les impuretés demeurent dans la phase aqueuse acidifiée, qui est neutralisée à la chaux avant élimination comme déchet.
Production d’acide phosphorique à l’aide d’acides autres que l’acide sulfurique
La roche phosphatée peut être dissoute par plusieurs acides organiques et minéraux afin de produire de l’acide phosphorique.
Les procédés commerciaux de type nitrophosphate produisent un acide phosphorique contenant des nitrates ; ces procédés sont donc utilisés pour produire des engrais composés NP ou NPK.
Il est techniquement possible de produire un acide phosphorique pratiquement exempt de calcium ou de nitrates par des méthodes de séparation faisant intervenir l’extraction par solvant.
Un tel procédé, utilisant l’alcool amylique tertiaire comme solvant, a été développé en Finlande et décrit par Lounamaa [40] ; toutefois, aucune utilisation commerciale n’a été rapportée.
Plusieurs procédés utilisant l’acide chlorhydrique ont été développés ou brevetés, mais peu ont été utilisés commercialement. Les principales étapes d’un tel procédé sont les suivantes :
Dissolution de la roche phosphatée par l’acide chlorhydrique, ce qui donne une solution aqueuse de chlorure de calcium et d’acide phosphorique ;
Contact liquide-liquide en plusieurs étapes d’extraction par solvant afin d’obtenir une solution d’acide phosphorique substantiellement pur ;
Et concentration de l’acide afin d’obtenir 95 % de H₃PO₄, soit 69 % de P₂O₅.
Les matières premières et réactifs utilisés sont les suivants :
Roche phosphatée, de toute qualité commerciale. Le taux de récupération du P₂O₅ est supérieur à 98 %.
Acide chlorhydrique pour l’attaque acide, pouvant être utilisé sous forme d’une solution à 20 % de HCl ou plus, ou sous forme gazeuse en combinant absorption et réaction. Pour des raisons économiques, l’acide concentré est préféré, car la majeure partie de l’eau accompagnant l’acide doit être évaporée à une étape ultérieure du procédé. La consommation de HCl dépend de la composition de la roche.
Solvant. Plusieurs solvants peuvent être utilisés pour l’extraction. Les solvants préférés sont l’alcool isoamylique technique, IM, le n-butanol, ou un mélange des deux. L’appoint de solvant est de 4 kg par tonne de P₂O₅.
Eau de procédé.
Réactifs auxiliaires. Selon le type de roche et la méthode de séparation du résidu insoluble de la liqueur de dissolution, de petites quantités d’adjuvants de filtration ou d’agents floculants peuvent être nécessaires.
Dissolution et séparation mécanique du résidu insoluble
La dissolution de la roche phosphatée correspond essentiellement à la décomposition de la fluoroapatite par HCl selon l’équation suivante :
Ca₁₀F₂(PO₄)₆ + 20 HCl → 2HF + 6H₃PO₄ + 10CaCl₂
D’autres constituants solubles dans l’acide présents dans la roche, tels que CaCO₃, se décomposent simultanément.
La roche est dissoute par l’acide chlorhydrique. Le résidu insoluble représente un faible pourcentage de l’alimentation en roche et se compose principalement de silice, de silicates et de matière organique insoluble.
La matière insoluble peut être séparée de la liqueur de dissolution par filtration, suivie du lavage du gâteau, ou par sédimentation dans un épaississeur, suivie d’un lavage par décantation à contre-courant du sédiment.
Le choix de la méthode appropriée de séparation des solides dépend de la nature du résidu insoluble et de considérations économiques. La liqueur de dissolution est envoyée vers la section suivante.
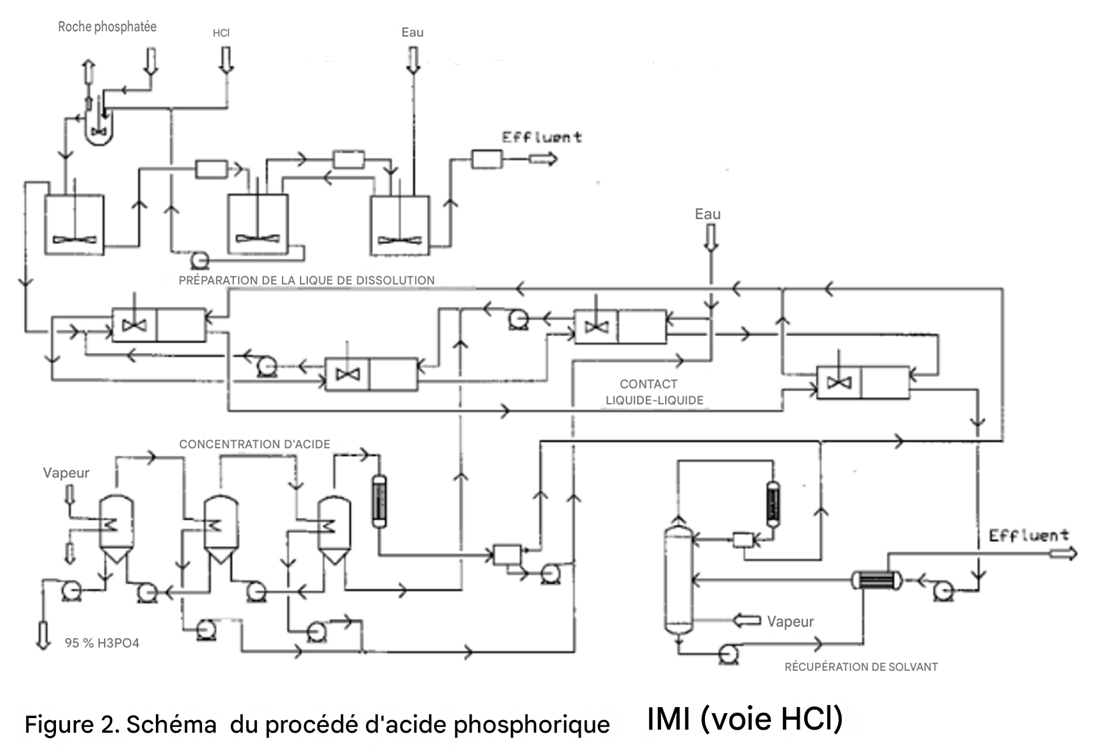
La figure 2 présente un schéma de procédé typique de production d’acide phosphorique par la voie HCl.
Contact liquide-liquide
Cette étape comprend plusieurs opérations : extraction, purification, lavage et stripping.
Extraction
L’extraction est réalisée par contact à contre-courant de la liqueur de dissolution avec le solvant sélectionné.
L’acide phosphorique est transféré sélectivement de la liqueur aqueuse de dissolution vers la phase solvant organique ; l’extrait obtenu ainsi que la saumure de chlorure de calcium, ou raffinat, contiennent pratiquement toutes les impuretés, telles que le fluor et le fer.
Purification
L’extrait de solvant, qui contient de faibles quantités de Ca²⁺ et quelques autres impuretés, est purifié davantage par contact à contre-courant avec une phase aqueuse.
Lavage
L’acide de l’extrait purifié est transféré dans l’eau. Le solvant quittant cette étape est pratiquement exempt d’acide.
Stripping
Le courant de solvant exempt d’acide extrait les acides résiduels présents dans le raffinat et est recyclé vers l’extraction. La saumure usée de chlorure de calcium est strippée à la vapeur afin de récupérer tout solvant dissous.
Concentration de l’acide
L’acide aqueux dilué provenant du lavage est constitué d’une solution aqueuse de H₃PO₄, de HCl et d’une certaine quantité de solvant dissous.
Cette solution est concentrée jusqu’à 95 % de H₃PO₄, qui constitue le produit final.
La séparation de H₃PO₄ des autres constituants de la solution est essentiellement une opération de distillation ; cela permet la récupération complète de la faible quantité de solvant dissoute dans la phase aqueuse lors du lavage, ainsi que du HCl, tous deux maintenus en circuit fermé dans le procédé.
L’exigence principale de cette opération est l’économie de chaleur, et un évaporateur à effets multiples est utilisé pour y parvenir.
La quantité de vapeur utilisée est inférieure à 0,5 tonne par tonne d’eau évaporée.
L’absence totale de solides dissous dans la solution à concentrer permet de maintenir des coefficients de transfert thermique élevés.
Tous les flux volatils du procédé sont recyclés vers les étapes précédentes du procédé.
Récupération du solvant à partir de la saumure usée de chlorure de calcium
La saumure résiduelle quittant l’étape de stripping contient une faible quantité de solvant dissous, qui doit être récupérée pour des raisons économiques.
Les solvants utilisés forment un azéotrope avec l’eau lors de la rectification ; le système le plus simple à appliquer est donc le stripping à la vapeur.
Les coûts de cette opération sont réduits par la récupération de chaleur à partir de la saumure sortant du système.
Le solvant récupéré est recyclé vers la section de contact liquide-liquide, et la saumure est rejetée.
Matériaux de construction
Dans les parties du procédé où le solvant est présent, des matériaux de construction résistants à l’acide et également résistants au solvant sont nécessaires.
Ces parties comprennent la section de contact liquide-liquide ainsi qu’une partie des sections où l’acide est concentré et où le solvant est récupéré à partir de la saumure.
L’acier revêtu de caoutchouc est le matériau le moins coûteux pour la dissolution et la séparation mécanique du résidu insoluble.
Dans la section de contact liquide-liquide, le polychlorure de vinyle rigide, PVC, convient.
Dans les parties du système fonctionnant à température élevée, le graphite imperméable peut être utilisé pour les échangeurs de chaleur.
D’autres matériaux de construction comprennent les résines thermodurcissables et l’acier revêtu.
Qualité de l’acide phosphorique obtenu par la voie HCl
L’acide phosphorique produit par la voie chlorhydrique est beaucoup plus propre que l’acide obtenu par voie humide, et son analyse est similaire à celle de l’acide thermique. En effectuant de légers ajustements dans le procédé, il est possible d’obtenir un acide de qualité alimentaire. La composition de l’acide phosphorique par voie humide dépend de la roche utilisée comme matière première ; alors que, pour l’acide phosphorique produit par voie HCl, c’est pratiquement l’inverse.
Tableau 1. Comparaison de l’analyse de l’acide phosphorique produit par la voie HCl et de l’acide phosphorique obtenu par voie humide
Composants
Voie HCl, %
Voie humide, %
H₃PO₄
95
69–77
P₂O₅
69
50–56
Métaux lourds exprimés en équivalent Pb
0.002–0.01
0.5–1.5
CaO
0.008–0.04
0.014–0.35
Fe₂O₃
0.003–0.05
0.86–2.3
Al₂O₃
traces
0.3–2.45
Mg
traces
0.0–0.8
H₂SO₄
traces
1.0–5.6
SiO₂
traces
0.04–0.10
F
traces
0.25–1.10
Investissement en capital
L’investissement en capital requis pour une unité peut varier d’un site à l’autre. Toutefois, à des fins de comparaison, il convient de noter que le coût d’investissement du procédé HCl est environ 35 % plus élevé que celui du procédé humide standard à l’acide sulfurique, H₂SO₄, lorsque la production des acides est exclue.
Si le HCl était disponible comme coproduit d’un autre procédé, le coût d’investissement serait inférieur à celui d’une unité par voie humide incluant les installations de production de H₂SO₄. Cependant, lors de l’utilisation de HCl coproduit, la capacité de l’opération serait limitée par la quantité de HCl coproduit disponible.
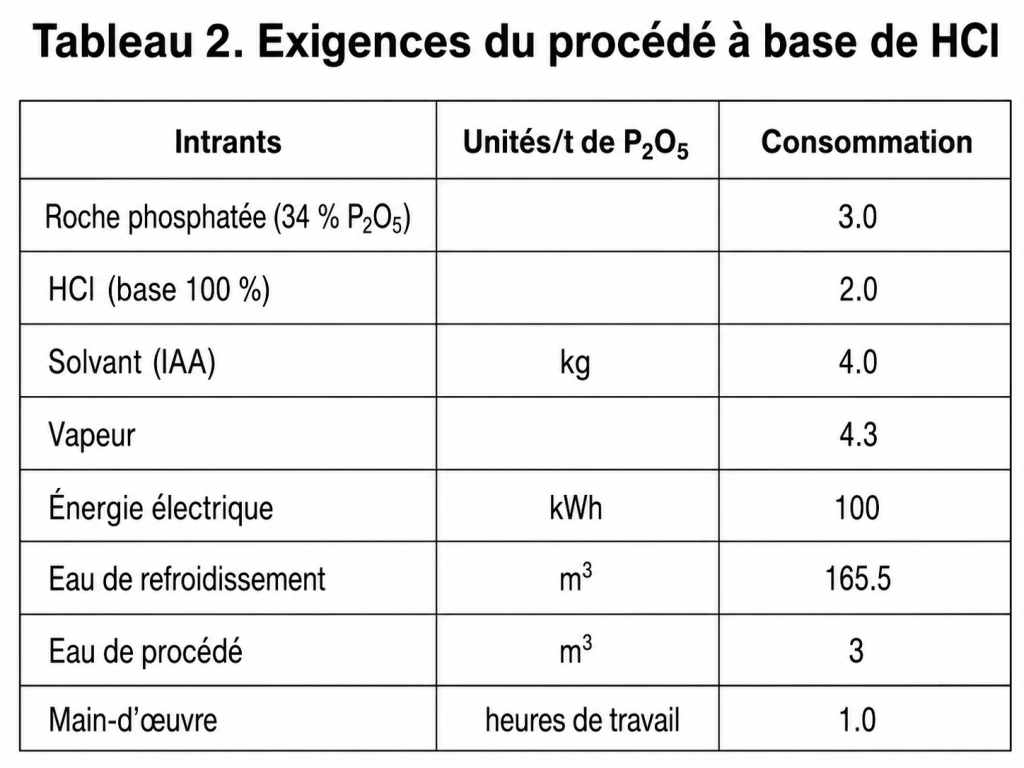
Exigences du procédé
Les coûts opératoires peuvent être estimés à partir des besoins du procédé, qui sont indiqués dans le tableau 2 pour une unité d’une capacité de 100 t/j.
L’élimination du CaCl₂ peut être difficile et coûteuse.
L’acide phosphorique produit par voie HCl présente certains inconvénients par rapport à l’acide par voie humide. Sa production n’est économique que dans les endroits où le HCl est disponible, ou bien là où il peut être produit à un prix modéré.
Le transport du HCl sous forme d’une solution aqueuse contenant éventuellement 33 % de HCl n’est possible que dans des conduites ou des wagons-citernes revêtus de caoutchouc, de PVC ou de matériaux similaires.
Cependant, l’acide phosphorique par voie HCl présente certains avantages par rapport à l’acide par voie humide. Contrairement à l’acide par voie humide, il ne contient pas de constituants générateurs d’entartrage, et sa composition ainsi que sa qualité sont pratiquement indépendantes du type de roche phosphatée utilisée.
L’acide superphosphorique, contenant 70 à 72 % de P₂O₅, peut être facilement produit à partir d’acide phosphorique voie HCl, sans procédé supplémentaire de purification.
Le HCl peut être utilisé lorsqu’il est disponible comme coproduit. Cela est important pour les pays producteurs de NaOH, lorsqu’il n’existe pas de marché captif pour le chlore produit simultanément.
L’acide chlorhydrique coproduit est parfois disponible à partir d’autres sources et peut même poser des problèmes d’élimination. Dans de tels cas, la production d’acide phosphorique par attaque acide de la roche phosphatée avec de l’acide chlorhydrique peut être avantageuse si la quantité de coproduit est suffisante pour permettre une échelle d’exploitation économique, et si l’élimination ou l’utilisation de la saumure de chlorure de calcium est économiquement faisable.
Une source potentielle intéressante d’acide chlorhydrique est la calcination et l’hydrolyse du chlorure de magnésium, selon l’équation suivante :
MgCl₂ + H₂O = 2HCl + MgO
L’oxyde de magnésium pourrait être utile pour la production de matériaux réfractaires.
Une autre source possible d’acide chlorhydrique provient de la production de phosphate de potassium à partir d’acide phosphorique et de chlorure de potassium.
À l’heure actuelle, les seules unités utilisant le procédé HCl sont des installations relativement petites, et la majeure partie du produit est utilisée pour fabriquer des phosphates industriels plutôt que des engrais.
Comme voie et procédé alternatifs, le phosphate dicalcique, DCP, peut être produit à partir de roche phosphatée et de HCl. Le DCP peut être utilisé pour l’alimentation animale ou être retraité avec de l’acide sulfurique afin de produire de l’acide phosphorique.
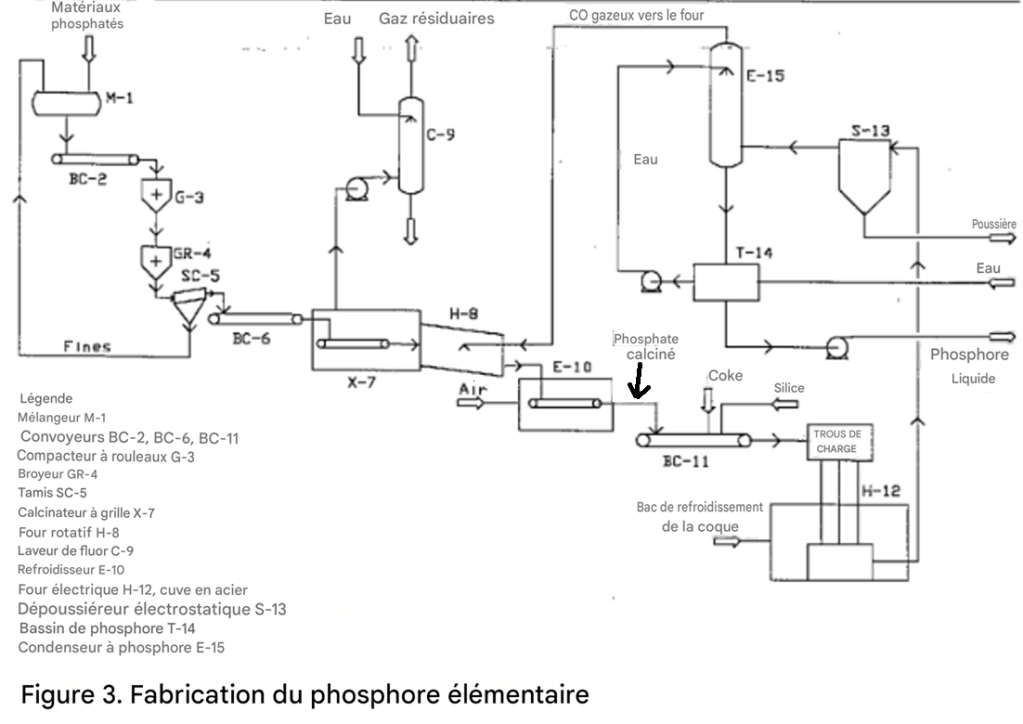
Production d’acide phosphorique par procédé au four électrique
La première étape de la production d’acide phosphorique par voie thermique consiste à produire du phosphore élémentaire dans un four électrique.
Des nodules phosphatés ou d’autres matériaux phosphatés en morceaux, des galets de silice et du coke sont mélangés puis introduits dans le four. Le courant électrique fourni au four par des électrodes en carbone ou en graphite fait fondre la roche et la silice, tandis que le carbone contenu dans le coke réduit le phosphate.
Un mélange de vapeur de phosphore et de monoxyde de carbone est soutiré en continu du four. Le phosphore est condensé sous forme liquide, puis converti en acide phosphorique dans une unité séparée, souvent située loin de l’unité de production de phosphore.
Des scories de silicate de calcium fondu et un composé fer-phosphore appelé ferrophosphore sont périodiquement soutirés du four.
L’équation suivante représente la réaction principale dans le four :
L’un des avantages du procédé au four est sa capacité à utiliser des roches phosphatées de faible teneur, à condition que l’impureté principale soit la silice. L’oxyde de fer et l’alumine sont beaucoup moins problématiques dans le procédé au four que dans le procédé par voie humide.
Des roches phosphatées siliceuses contenant environ 24 % de P₂O₅ sont utilisées dans plusieurs unités ; ce type de roche peut être obtenu à très faible coût dans certaines régions. Une roche contenant jusqu’à 7 % de Al₂O₃ est acceptable.
Si une roche en morceaux ou en galets, de granulométrie appropriée, environ 0,6 à 4,0 cm, et résistante à la décrépitation lors du chauffage est disponible, le coût d’agglomération de la charge peut être évité.
Cependant, une telle roche est rarement disponible ; par conséquent, la roche est généralement agglomérée puis calcinée ou frittée avant son introduction dans le four. Le monoxyde de carbone, coproduit du four, est le combustible habituel utilisé pour la calcination. Malgré cela, cette étape reste coûteuse.
La récupération du P₂O₅ sous forme de phosphore élémentaire se situe généralement entre 86 et 92 % de la quantité introduite dans le four. La perte de P₂O₅ dans les scories est d’environ 3 %.
Entre 2 et 8 % du P₂O₅ introduit est récupéré sous forme de ferrophosphore, lequel contient environ 23 % de phosphore, 70 % de fer, ainsi que de petites quantités de manganèse, de silicium et d’autres éléments métalliques, selon la composition de la charge.
La quantité de ferrophosphore formée dépend de la teneur en oxyde de fer de la charge. Le ferrophosphore est vendu à l’industrie sidérurgique, mais le revenu généré ne compense que partiellement la perte de production de phosphore.
Parmi le phosphore récupéré sous forme de phosphore élémentaire, environ 5 % se trouve sous forme de boues, même après une série d’étapes de décantation destinées à séparer les boues du phosphore propre.
Ce phosphore contenu dans les boues peut être récupéré par combustion séparée afin de produire un acide phosphorique impur, par distillation, ou par déshydratation puis retour au four.
Les principaux inconvénients du procédé au four sont le coût d’investissement relativement élevé de l’unité et la rareté des sites où l’électricité à faible coût est disponible.
Pour cette raison, le procédé au four électrique est utilisé presque exclusivement pour produire du phosphore et de l’acide phosphorique destinés aux produits chimiques industriels, aux insecticides, aux détergents et aux additifs alimentaires ou pour l’alimentation animale.
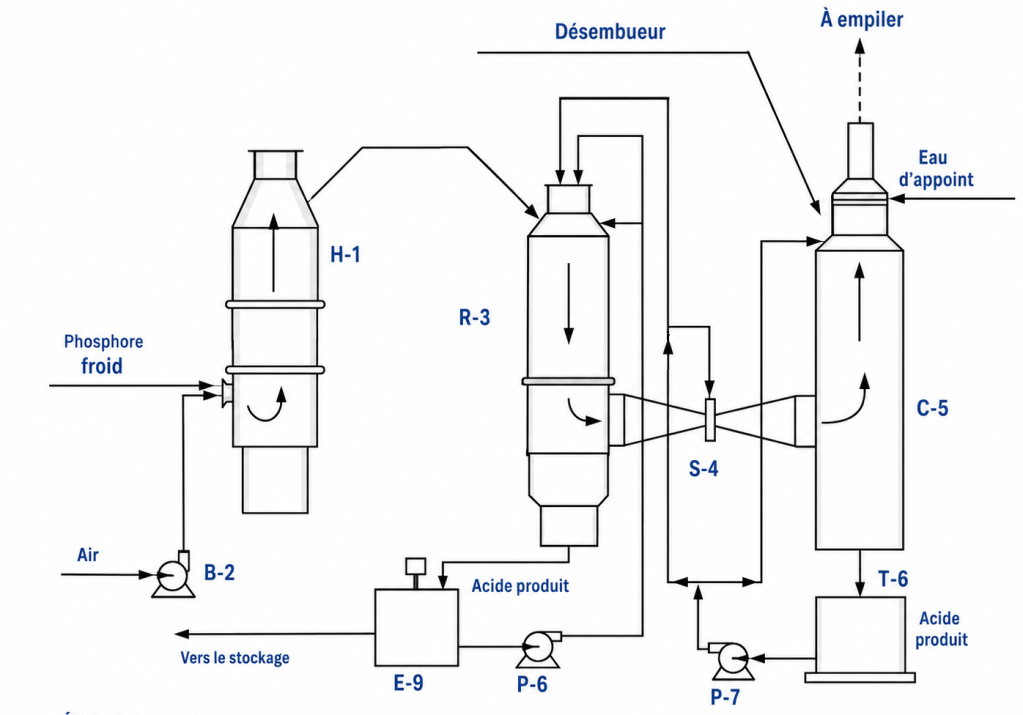
La production d’acide phosphorique à partir de phosphore élémentaire est relativement simple. Elle est réalisée par combustion du phosphore élémentaire liquide dans l’air, puis hydratation du P₂O₅ obtenu en H₃PO₄.
Un schéma d’une unité typique est présenté dans la figure 4. L’ensemble des équipements de procédé est fabriqué en acier inoxydable, généralement de type 316.
Figure 4. Fabrication d’acide phosphorique à partir de phosphore élémentaire
La réaction globale est la suivante :
P₄ + 5O₂ + 6H₂O → 4H₃PO₄
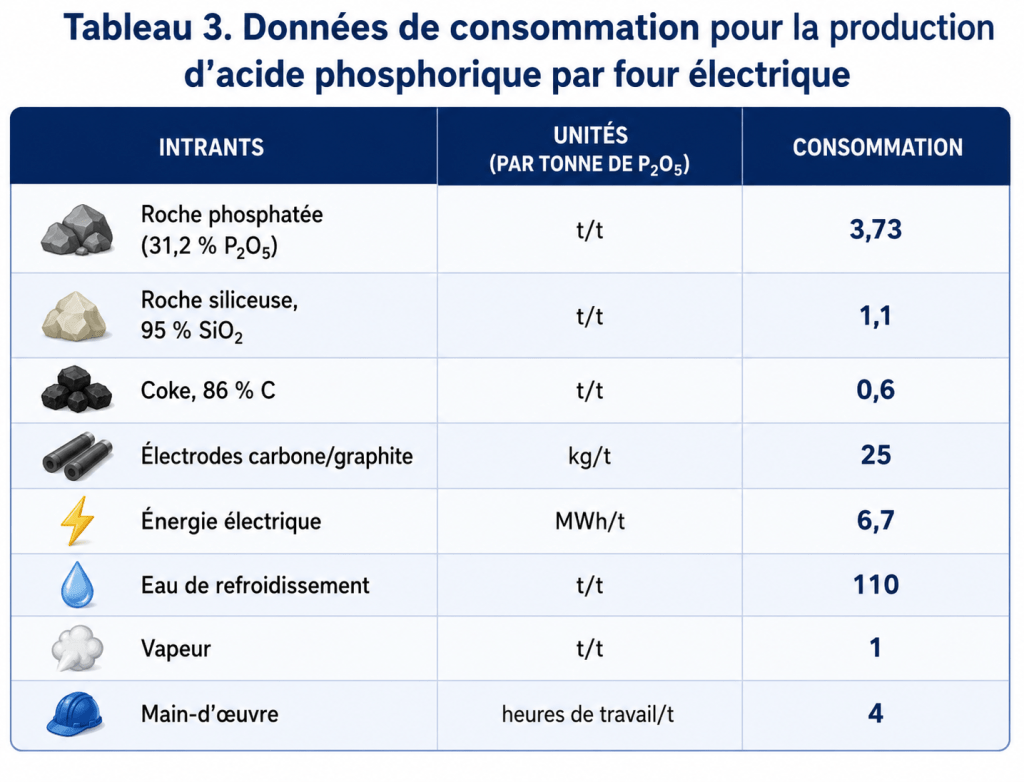
Les besoins typiques du procédé par tonne de P₂O₅ produite sous forme d’acide phosphorique, en supposant un rendement global de 86 % pour une unité d’environ 100 000 tonnes de P₂O₅ par an, sont indiqués dans le tableau 3.
La consommation de combustible est négligeable, environ 1,2 million de kcal, selon l’efficacité d’utilisation du monoxyde de carbone coproduit.
Si une roche phosphatée satisfaisante est disponible pour une utilisation sans agglomération ni calcination, le coût de l’unité serait inférieur de 25 à 30 %, les coûts de maintenance et de main-d’œuvre seraient également inférieurs de 25 à 30 %, et les besoins en combustible seraient pratiquement éliminés.
Le monoxyde de carbone coproduit par le four serait plus que suffisant pour le séchage du coke, de la silice et de la roche.
Certaines roches ont été utilisées avec succès dans des fours électriques sans agglomération, calcination ni frittage : il s’agit notamment du galet phosphaté de Floride, plus de 6 mm, de la roche dure de Ronda et de la roche dure du Montana, concassée et criblée.
L’utilisation de roche non calcinée peut augmenter la consommation d’énergie électrique dans le four jusqu’à 10 %, selon la teneur en CO₂ et en eau combinée.
La Tennessee Valley Authority, TVA, a commencé le développement du procédé au four électrique pour produire des engrais phosphatés en 1933 et a produit du phosphore et de l’acide phosphorique de 1934 à 1977.
À une époque, cinq fours étaient en fonctionnement. En 1977, l’exploitation de tous les fours a été arrêtée par la TVA, car le procédé ne pouvait plus concurrencer le procédé par voie humide pour la production d’engrais.
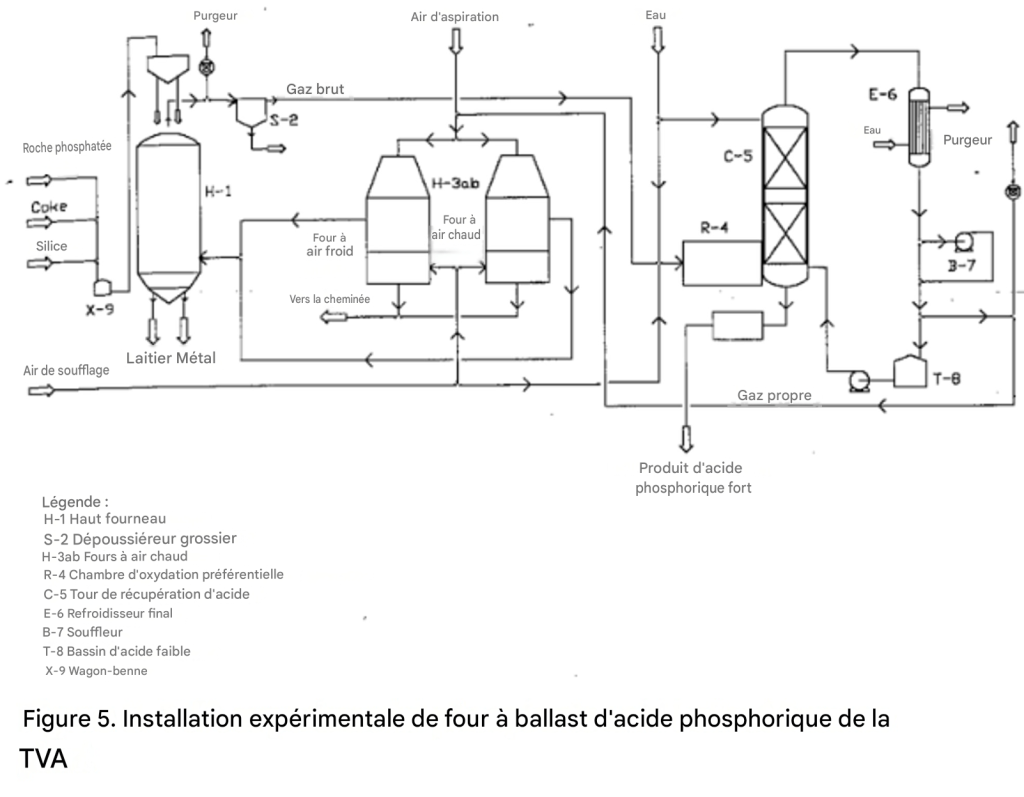
Production d’acide phosphorique par procédé au haut-fourneau
Un schéma de procédé d’une unité pilote de la TVA pour la production d’acide phosphorique par procédé au haut-fourneau est présenté dans la figure 5.
Figure 5. Unité pilote expérimentale de la TVA pour la production d’acide phosphorique par procédé au haut-fourneau
L’échelle de l’unité pilote était d’environ 1 tonne de P₂O₅ par jour [43].
En général, le procédé au haut-fourneau diffère du procédé au four électrique sur les points suivants :
Le coke est utilisé à la fois comme combustible et comme agent de réduction du phosphore. Le besoin estimé en coke pour une grande unité est de 2,5 tonnes par tonne de P₂O₅ récupéré sous forme d’acide phosphorique, en tenant compte des pertes en ferrophosphore.
Environ 0,6 tonne de coke est consommée pour la réduction du P₂O₅ en phosphore, et le reste produit de la chaleur par combustion avec de l’air préchauffé pour former du monoxyde de carbone.
Comme dans le cas du four électrique, la charge — roche phosphatée, coke et silice — doit se présenter sous forme de morceaux ou d’agglomérés. Toutefois, il n’est pas nécessaire de calciner ni de sécher la charge, car les gaz ascendants dans la cuve du haut-fourneau fournissent suffisamment de chaleur à cet effet.
Le gaz provenant du four contient environ 37 % de CO et 1,0 à 1,5 % de P₄ en volume. Le reste est principalement constitué d’azote.
Bien que la récupération du phosphore élémentaire par refroidissement et condensation soit possible, il serait difficile d’en récupérer un pourcentage élevé en raison de sa faible concentration dans le gaz.
Dans l’unité pilote de la TVA, l’acide phosphorique était récupéré après oxydation préférentielle du phosphore contenu dans le gaz par l’air.
Le gaz restant après récupération de l’acide phosphorique contient environ 34 % de CO, 1 à 2 % de O₂, et le reste est constitué de N₂, sur base sèche.
Environ 40 % de ce gaz peut être utilisé avantageusement pour préchauffer l’air destiné au haut-fourneau. Le reste serait disponible pour d’autres usages.
L’utilisation du haut-fourneau pour produire de l’acide phosphorique destiné aux engrais semble actuellement peu prometteuse en raison du coût élevé du coke. Toutefois, avec certaines améliorations, elle pourrait être envisagée dans certaines circonstances [44].
Comme le procédé au four électrique, il peut utiliser une roche siliceuse de faible teneur présentant une teneur modérément élevée en alumine et en oxyde de fer.
Références
Sanders, M.D. 1968. ‘Recovery of Fluorides as By-products’. IN Phosphoric acid, A.V. Slack (Ed.) pp765-778, Marcel Dekker Inc., New York, NY, USA.
‘Production of Synthetic Fluorspar from Waste Fluosilic Acid’. 1976. Paper No 22. ISMA Technical Conference, The Hague, Netherlands.
Lounamaa, N. and L. Niinimaka. 1971. ‘Typpi Oy’s Solvent Extraction Process for Producing Compound Fertilizers’, Paper No. ID/WG99/20, Presented at the Second Interregional Fertilizer Symposium (UNIDO), Kiev, USSR.
‘Phosphoric Acid Production from Hydrochloric Acid.’ 1987. Negev Phosphates Ltd. Marketing Division, Israel.
Hignett, T. P. 1948. ‘Development of Blast Furnace Process for Production of Phosphoric Acid,’ Chemical Engineering Progress, 44(10): 753-764; 44(11):821-832; and 44(12):895-904.
Hignett, T. P. 1968. ‘Use of the Blast Furnace for Production of Phosphoric Acid,’ Phosphorus and Potassium, 36:20-21,24.
187, (1979), Uranium Recovery from Phosphoric Acid (A Process Engineering Review), A P Kouloheris
201, (1981), From Wet Crude Phosphoric Acid to High Purity Products, A Davister, G Martin
587, (2006), Phosphogypsum Management and Opportunities for Use, J Hilton
668, (2010), The Phosphate Life-Cycle: Rethinking the Options for a Finite Resource, J Hilton, A E Johnston, C J Dawson
744, (2014), History, Development, Status and Opportunities for Kiln Phosphoric Acid, T P Fowler
748, (2014), Management of Fluorine in Phosphoric Acid Production, B Van Massenhove, M G J A Collin, T Theys
752, (2014), Life Cycle Management of Phosphogypsum Stacks, G R Albarelli, B K Birky
766, (2015), Uranium Extraction from Phosphoric Acid: The Experience of Prayon, M G J A Collin
804, (2017), Phosphogypsum stacking: A new approach and case study, V Dardenne, J Peret and S Plainchamp
821, (2018), Approaches to improving the quality of phosphoric acid, T Henry
Becker, P. (1989) Phosphates and Phosphoric Acid: Raw Materials: Technology, and Economics of the Wet Process. Marcel Dekker, Inc., New York, NY, U.S.A.
Havelange, S. et al. (2022) Phosphoric Acid and Phosphates in Ullmann’s Encyclopaedia of Industrial Chemistry.
Slack, A.V. (1968). Phosphoric Acid (Part I and II). Marcel Dekker, Inc., New York, NY, U.S.A.
L’AGENCE INTERNATIONALE DE L’ÉNERGIE ATOMIQUE VIENNE, 2025
APERÇU GÉNÉRAL
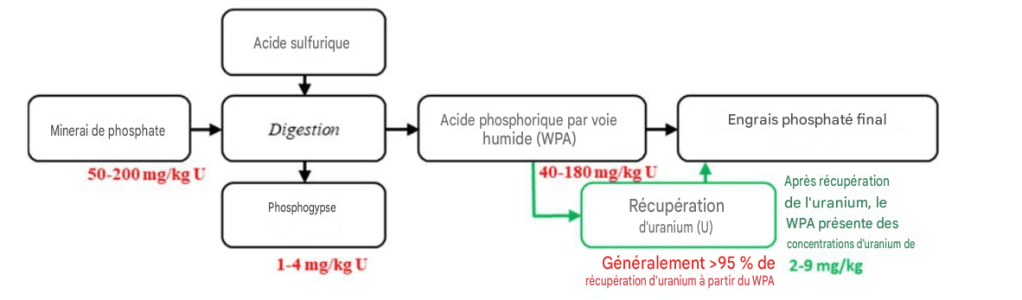
La plupart des minerais phosphatés sont d’origine sédimentaire, et c’est ce type de minerais phosphatés qui peut contenir des concentrations élevées en uranium, présentant typiquement des teneurs en uranium comprises entre 50 et 200 mg/kg [10]. Les grandes usines d’engrais phosphatés traitent entre 500 000 et 2 millions de tonnes de minerai phosphaté par an, ce qui représente un potentiel notable de récupération de l’uranium, même si celui-ci n’est présent qu’à l’état de traces dans les minerais phosphatés bruts traités. La Fig. 1, à gauche, montre un minerai phosphaté sédimentaire provenant de Floride, aux États-Unis, avant une concentration simple par flottation. Le minerai phosphaté concentré ou enrichi, également appelé concentré de roche phosphatée, issu du même minerai, est montré à la Fig. 2, à droite.
FIG. 2. Minerai de phosphate sédimentaire provenant de Floride, à gauche, et concentré obtenu à partir de ce minerai après enrichissement par flottation, à droite. Avec l’aimable autorisation de P. Zhang, Florida Polytechnic University.
Le minerai phosphaté est en grande partie insoluble et ne peut pas être directement utilisé comme engrais, sauf lorsque le sol est très acide, comme c’est le cas dans certaines régions tropicales. Le minerai phosphaté est donc transformé en engrais hydrosolubles afin que le phosphore puisse être absorbé par les plantes à travers leur système racinaire. Les usines d’acide phosphorique par voie humide — WPA, Wet Process Phosphoric Acid — constituent aujourd’hui le type d’usine d’engrais le plus courant en exploitation. Plus de 85 % des engrais phosphatés sont produits à partir de WPA, en utilisant de l’acide sulfurique pour l’attaque du minerai phosphaté.
Au cours du procédé WPA à l’acide sulfurique, représenté schématiquement à la Fig. 2, environ 80 à 90 % de l’uranium présent dans le minerai phosphaté est transféré vers l’acide phosphorique liquide WPA, à partir duquel il a historiquement été récupéré au moyen de différentes méthodes d’extraction par solvant. Les 10 à 20 % restants de l’uranium présent dans le minerai phosphaté sont généralement transférés vers le phosphogypse, un sous-produit solide du procédé de production de WPA.
Le phosphogypse peut avoir des usages bénéfiques, tels que l’application aux sols ou l’utilisation comme matériau de construction. Toutefois, ces usages sont dans certains cas limités par la présence d’isotopes radioactifs tels que le ²²⁶Ra ainsi que d’autres éléments traces toxiques. Pour traiter 1 tonne de minerai phosphaté, environ 0,6 tonne d’acide sulfurique concentré est utilisée. Le procédé génère ensuite approximativement 0,4 tonne de WPA de qualité marchande et 1,2 tonne de phosphogypse [11]. Historiquement, l’uranium était récupéré à partir du WPA de qualité marchande avant une concentration supplémentaire.
FIG. 3. Schéma simplifié du procédé de production d’acide phosphorique par voie humide (WPA) avec récupération de l’uranium, tel qu’il était historiquement mis en œuvre.
À certaines périodes où les prix de l’uranium étaient relativement élevés, la récupération des métaux lourds pendant la production d’engrais minéraux était rentable sur le plan financier pour les entreprises productrices de WPA. Les principaux facteurs qui déterminent la rentabilité économique sont, sans ordre particulier et sans tenir compte des réglementations environnementales ni des considérations stratégiques [9] :
la concentration en uranium dans le minerai phosphaté : plus elle est élevée, mieux c’est ;
la capacité de l’usine de traitement du minerai phosphaté : plus elle est grande, mieux c’est ;
le procédé appliqué pour la récupération de l’uranium : moins il est coûteux, mieux c’est.
Les considérations stratégiques comprennent l’amélioration de la sécurité d’approvisionnement en uranium — approvisionnement national — et, par conséquent, la diminution de la dépendance vis-à-vis des importations d’uranium, les économies en devises étrangères, l’acquisition de technologie et la création d’emplois. Il est à noter que la majeure partie de l’uranium est transférée vers le produit fertilisant final, et que cet uranium peut provoquer une contamination des sols et des eaux souterraines. Il existe une préoccupation particulière concernant l’accumulation potentielle de l’uranium, qui peut compromettre la fertilité des sols, migrer vers les eaux souterraines, voire être absorbé par les cultures et ingéré par les membres du public [12–14].
La motivation à l’origine des premières opérations de récupération de l’uranium à partir des phosphates était d’abord stratégique, dans une première période allant des années 1950 aux années 1960, lorsque les pays exploraient différentes sources de minerais uranifères ; elle est ensuite devenue commerciale, dans une seconde période allant des années 1970 à la fin des années 1990.
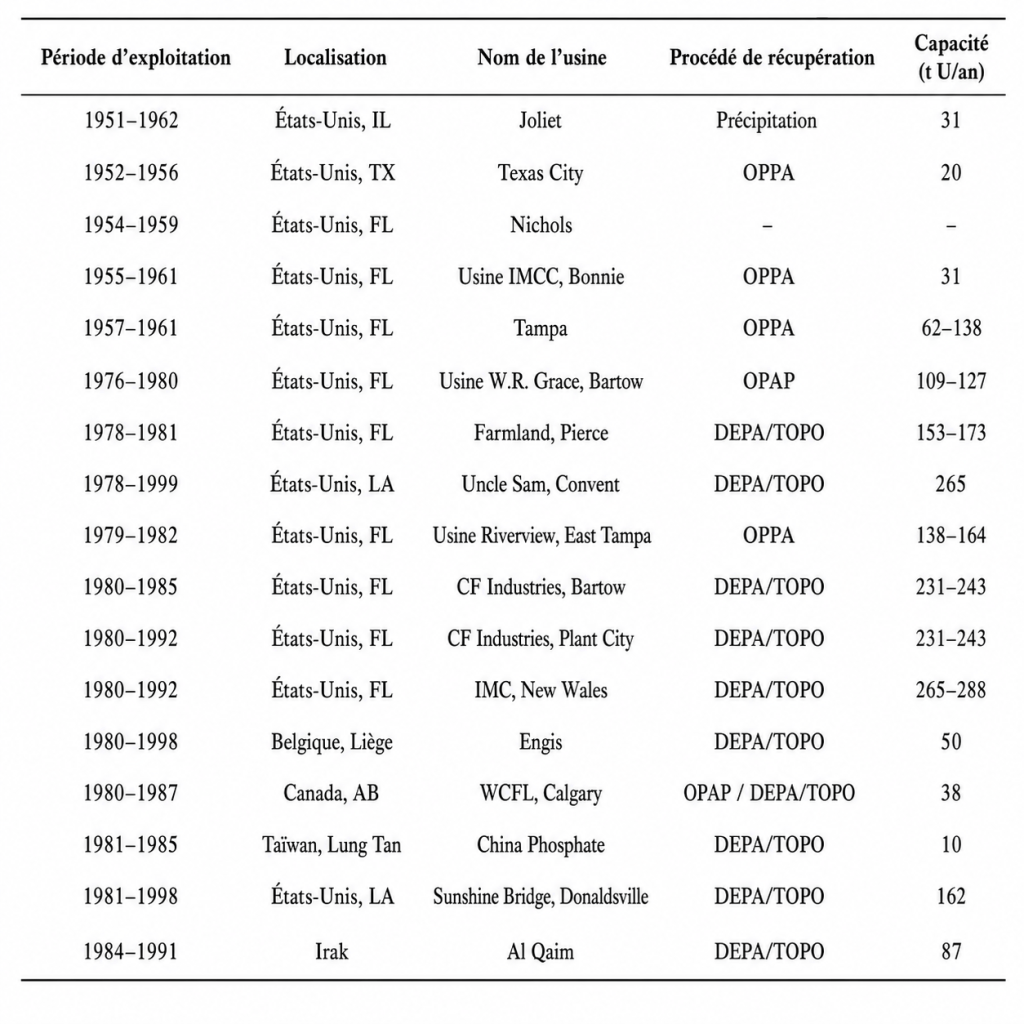
Le Tableau 2 résume les anciennes activités commerciales de récupération de l’uranium menées en Belgique, au Canada, en Irak, à Taïwan et aux États-Unis. Presque toutes les usines utilisaient l’extraction par solvant, et les différents solvants employés pour récupérer l’uranium à partir de la phase inorganique ont donné leur nom aux procédés correspondants. Le solvant le plus largement utilisé était le procédé DEPA–TOPO, également connu sous le nom de procédé ORNL, d’après l’Oak Ridge National Laboratory aux États-Unis, où la technique a été initialement mise au point. TOPO signifie oxyde de tri-n-octylphosphine, et DEPA désigne l’acide bis(2-éthylhexyl)phosphorique.
TABLEAU 2. HISTORIQUE DE RÉCUPÉRATION DE L’URANIUM À PARTIR DU MINERAI PHOSPHATÉ, PAR RÉGION ET PAR USINE.
PROCÉDÉ D’EXTRACTION PAR SOLVANT ÉPROUVÉ COMMERCIALEMENT
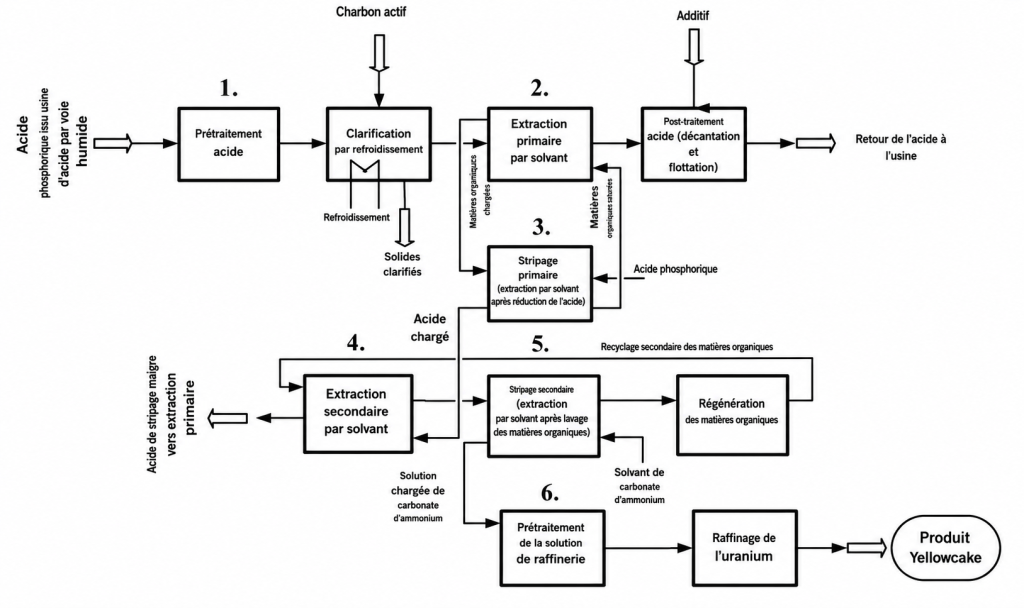
Les procédés d’extraction par solvant ont constitué les méthodes de récupération de l’uranium déployées avec succès pour extraire l’uranium du WPA liquide à l’échelle industrielle commerciale. Beltrami et al. [15], Bunus [10] et Singh et al. [16] présentent des revues de littérature scientifique sur les procédés utilisés. La Fig. 3 fournit un bref aperçu du procédé d’extraction par solvant en deux cycles — DEPA/TOPO dans le Tableau 2 — qui s’est révélé être le plus performant par le passé.
FIG. 4. Procédé d’extraction par solvant à deux cycles, utilisé historiquement pour la récupération commerciale de l’uranium à partir de l’acide phosphorique par voie humide (WPA).
Les principes fondamentaux du procédé DEPA/TOPO présenté à la Fig. 4 sont les suivants :
Prétraitement acide : après refroidissement et, occasionnellement, élimination des matières organiques — ce qui entraîne une décoloration — l’acide phosphorique provenant du filtre, contenant 25 à 30 % de P₂O₅, est clarifié par élimination des solides.
Extraction primaire par solvant : l’acide clarifié est introduit dans un système mélangeur-décanteur à contre-courant contenant le solvant DEPA/TOPO dissous dans du kérosène. L’uranium est transféré vers la phase solvant, ou phase dite organique chargée. Le raffinat, parfois appelé acide phosphorique pauvre ou appauvri, est renvoyé vers l’usine d’acide phosphorique.
Désextraction primaire : un acide contenant un agent réducteur est mis en contact avec la solution organique chargée afin de favoriser le passage de l’uranium de l’état U(VI) à l’état U(IV). Ensuite, dans un second système mélangeur-décanteur, l’uranium entre en contact avec de l’acide phosphorique très concentré. À ce stade, l’uranium est retiré de la quantité relativement importante de solvant organique et transféré vers un volume plus faible d’acide de désextraction, c’est-à-dire l’acide principal de désextraction chargé. Ce procédé entraîne une augmentation significative de la concentration en uranium par rapport à l’étape précédente. Des facteurs de concentration pouvant atteindre 20 sont généralement observés à ce niveau.
Extraction secondaire par solvant : l’uranium est oxydé dans l’acide principal de désextraction chargé afin de le ramener à la forme U(VI). L’uranium concentré est transféré vers la phase solvant et encore davantage concentré afin de générer une phase appelée organique secondaire chargée, lorsque l’acide de désextraction entre en contact avec le solvant DEPA/TOPO dans un système mélangeur-décanteur.
Désextraction secondaire : dans un système mélangeur-décanteur, une solution alcaline entre en contact avec la solution organique secondaire chargée contenant l’uranium. Au cours de ce procédé, l’uranium est libéré du solvant organique et transféré sous forme concentrée vers la solution alcaline. Une solution uranifère acide est ensuite produite en traitant la solution de désextraction secondaire afin d’éliminer les alcalis et de la neutraliser. À ce stade, la teneur en uranium est d’environ 20 g/L.
Raffinage : la solution acide contenant l’uranium est ensuite soumise à un traitement supplémentaire afin de produire du peroxyde d’uranyle, du diuranate d’ammonium — ADU — ou du tricarbonate d’uranyle ammonium — AUT — qui est ensuite épaissi, lavé, séché et calciné afin d’obtenir de l’U₃O₈ ou yellowcake.
La Fig. 5 montre l’usine d’acide phosphorique de New Wales de la société International Minerals and Chemical Corporation (IMC) Phosphates — devenue aujourd’hui Mosaic Fertilizer Company — ainsi que les installations d’extraction des premier et second cycles, exploitées en Floride, aux États-Unis, entre 1980 et 1992.
FIG. 5. Unité de récupération de l’uranium intégrée à l’usine d’engrais phosphatés IMC New Wales, située en Floride, aux États-Unis — avec l’aimable autorisation de V. Astley, Dr. Phosphate Inc.
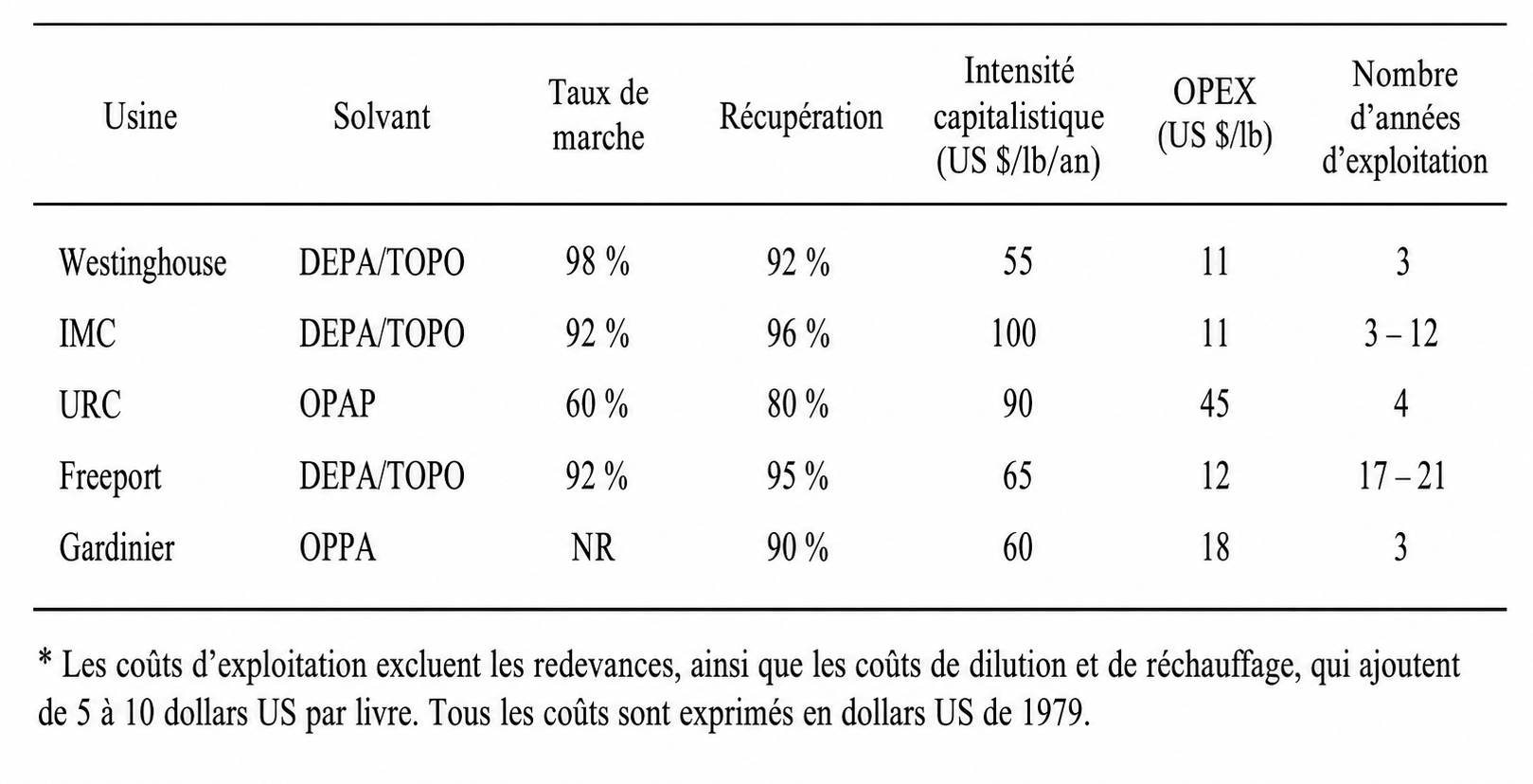
RETOURS D’EXPÉRIENCE ET ESTIMATIONS DES COÛTS DE LA RÉCUPÉRATION HISTORIQUE DE L’URANIUM
Le succès de l’extraction par solvant dans les années 1980 et 1990 a été porté par des intérêts commerciaux. D’excellents taux de récupération de l’uranium, dépassant 92 à 95 %, ainsi que le faible impact sur le cœur de métier des engrais, ont permis la récupération de l’uranium à l’échelle industrielle. Lorsque, à la fin des années 1990, la récupération de l’uranium a finalement cessé dans les usines de Floride et de Louisiane, les exploitants ont effectivement reconnu que la qualité de l’acide phosphorique avait été légèrement réduite.
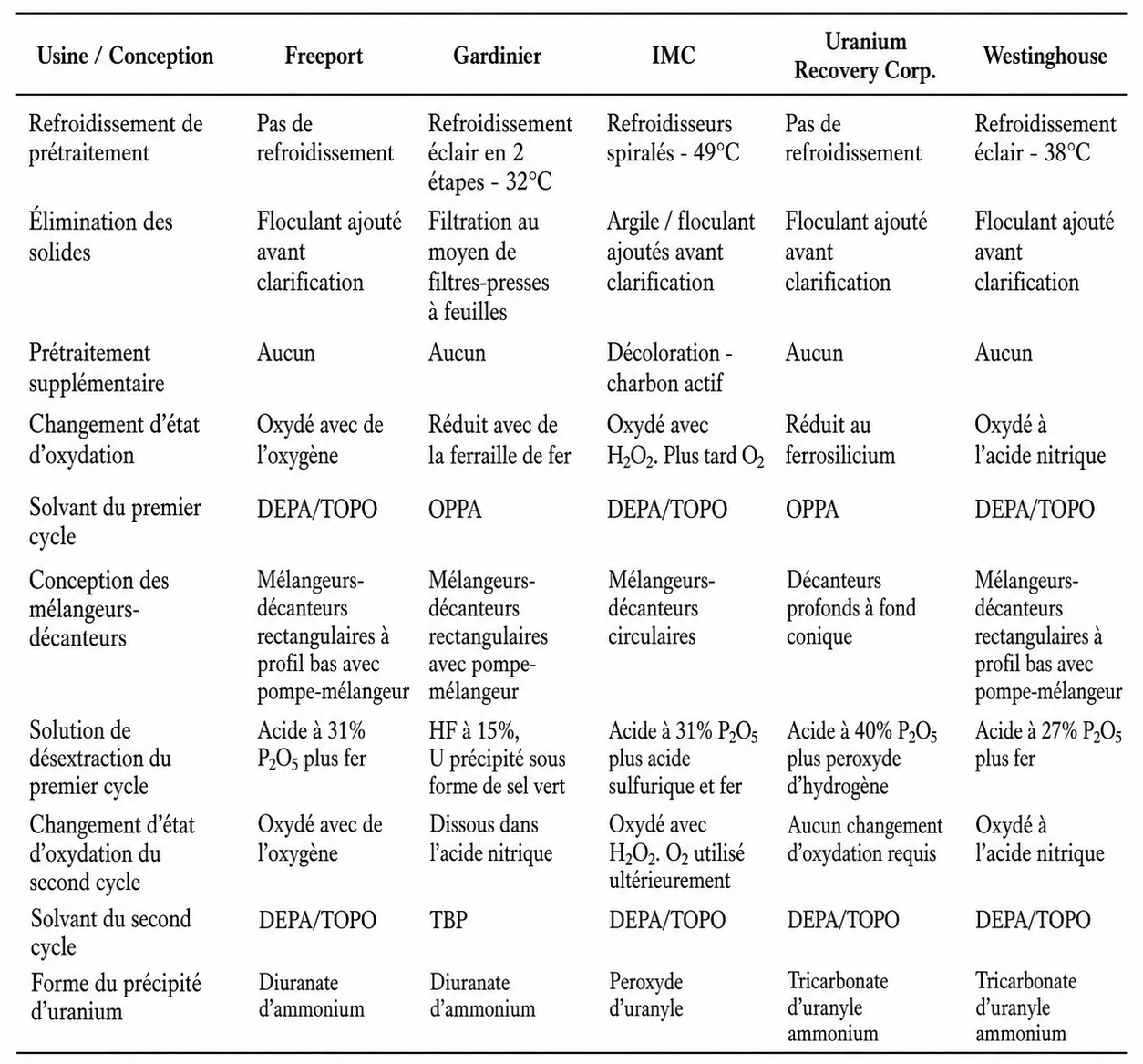
Les usines de récupération de l’uranium les plus performantes utilisaient le même solvant — DEPA/TOPO — et chacune fonctionnait avec de l’acide phosphorique produit par le procédé dihydrate. L’acide contenait typiquement de l’uranium à une concentration d’environ 0,5 kg/t. Certaines des différences les plus significatives dans la conception des installations sont résumées dans le Tableau 3.
TABLEAU 3. DIFFÉRENCES ENTRE LES ANCIENNES USINES COMMERCIALES METTANT EN ŒUVRE L’EXTRACTION PAR SOLVANT DEPA/TOPO.
Malgré ces différences, les usines présentant les volumes de production les plus élevés ont atteint de très hauts niveaux d’efficacité opérationnelle et de récupération de l’uranium. Les paramètres économiques sous-jacents des usines les plus efficaces étaient globalement similaires. Le Tableau 4 présente certaines plages de données relatives à l’exploitation et aux coûts pour ces mêmes usines. L’hypothèse de calcul des coûts figurant dans le tableau est fondée sur une teneur en uranium dans l’acide d’environ 0,5 kg/t.
TABLEAU 4. Données économiques relatives à l’historique de la récupération de l’uranium à partir des installations d’acide phosphorique (dollars US de 1979)*
Il existait d’autres différences importantes de performance. Pour les trois usines ayant utilisé le DEPA/TOPO, les pertes de solvant auraient par exemple représenté des coûts allant de 2 à 5,50 dollars US par livre de U₃O₈. Les coûts de prétraitement variaient de 0,20 dollar US à plus de 4,00 dollars US par livre de U₃O₈. Les coûts combinés des pertes de solvant et du prétraitement variaient de 2,50 dollars US à plus de 8,00 dollars US par livre de U₃O₈.
Les pertes moyennes de solvant vers le raffinat variaient de 5 à 100 mg/kg. Les pertes de P₂O₅ variaient de moins de 0,1 % à environ 1,0 %. Les coefficients effectifs de désextraction variaient de 15 à 150 et ne présentaient aucune corrélation significative avec la concentration en P₂O₅ de l’acide de désextraction. Les pertes de solvant dues au nettoyage des décanteurs variaient de moins de 0,15 à plus de 0,5 kg/t de P₂O₅ traité. Les effets en aval du procédé de récupération de l’uranium variaient de nuls à plusieurs millions de dollars par an, principalement en raison d’une diminution de la durée de vie des revêtements en caoutchouc.
PERTINENCE DE L’URANIUM ISSU DES PHOSPHATES AUX ÉTATS-UNIS D’AMÉRIQUE
La récupération de l’uranium à partir des phosphates aux États-Unis a été particulièrement remarquable. À son apogée, cette technique a représenté jusqu’à 20 % de la production nationale d’uranium dans les années 1980, avant que la baisse des prix de l’uranium ne rende cette méthode économiquement irréalisable. Aux États-Unis, la récupération historique et prospective de l’uranium à partir des phosphates est comparée aux importations historiques d’uranium et à l’extraction minière nationale à la Fig. 6.
FIG. 6. Historique des Importationsd’uranium des États-Unis, des activités d’extraction minière, de la récupération de l’uranium à partir des phosphatesainsi que sa récupération prospective
Les États-Unis, comme de nombreux autres pays qui dépendent fortement de l’énergie nucléaire pour produire de l’électricité, importent désormais la majeure partie de l’uranium dont ils ont besoin depuis l’étranger. Il est intéressant de noter que, depuis les années 1990, la quantité d’uranium susceptible d’être récupérée à partir des phosphates comme sous-produit de la fabrication des engrais a eu le potentiel de dépasser la quantité extraite sur le territoire national. À cet égard, comme le suggèrent Steiner et al. [17], les préoccupations actuelles concernant la sécurité d’approvisionnement du pays en uranium pourraient effectivement « rendre de nouveau attractive la récupération de l’uranium à partir des phosphates ».
Références
[10] BUNUS, F.T., Uranium and Rare Earth Recovery from Phosphate Fertilizer Industry by Solvent Extraction, Mineral Processing and Extractive Metallurgy Review 21 (2000) 381-478, https://doi.org/10.1080/08827500008914174.
[11] BILAL, E., BELLEFQIH, H., BOURGIER, V., MAZOUZ, H., DUMITRAS, D.G., BARD, F., LABORDE, M., CASPAR, J.P., GUIHOT, B., IATAN, E.L., BOUNAKHLA, M., IANCU, M.A., MARINCEA, S., EASSKHRAOUI, M., LI, B., DIWA, R.R., RAMIREZ, J.D., CHERNYSH, Y., CHUBUR, V., ROUBIK, H., SCHMIDT, H., BENIAZZA, R., CANOVAS, C.R., NIETO, J.M., HANEKLAUS, N., Phosphogypsum circular economy considerations: A critical review from more than 65 storage sites worldwide, Journal of Cleaner Production 414 (2023) 137561, https://doi.org/10.1016/j.jclepro.2023.137561.
[12] BIGALKE, M., ULRICH, A., REHMUS, A., KELLER, A., Accumulation of cadmium and uranium in arable soils in Switzerland, Environmental Pollution 221 (2017) 85-93, https://doi.org/10.1016/j.envpol.2016.11.035.
[13] SCHNUG, E., LOTTERMOSER, B.G., Fertilizer-Derived Uranium and its Threat to Human Health, Environmental Science & Technology 47 (2013) 2433-2434, https://doi.org/10.1021/es4002357.
[14] INTERNATIONAL ATOMIC ENERGY AGENCY, The Environmental Behaviour of Uranium, Technical Reports Series No. 488, IAEA, Vienna (2023).
[15] BELTRAMI, D., COTE, G., MOKHTARI, H., COURTAUD, B., MOYER, B.A., CHAGNES, A., A Recovery of Uranium from Wet Phosphoric Acid by Solvent Extraction Processes, Chemical Reviews 114 (2014) 12002-12023, https://doi.org/10.1021/cr5001546.
[16] SINGH, D.K., MONDAL, S., CHAKRAVARTTY, J.K., Recovery of Uranium from Phosphoric Acid: A Review, Solvent Extraction and Ion Exchange 34 (2016) 201-225, https://doi.org/10.1080/07366299.2016.1169142.
[17] STEINER, G., GEISSLER, B., HANEKLAUS, N., Making Uranium Recovery from Phosphates Great Again? Environmental Science and Technology 54 (2020) 1287-1289, https://doi.org/10.1021/acs.est.9b07859.
Procédé hémihydrate-dihydrate avec filtration intermédiaire — HDH
Procédé dihydrate-hémihydrate — DHH
Ingénierie et construction de l’unité
Comme pour les procédés dihydrate, plusieurs procédés hémihydrate et systèmes combinés ont été développés par diverses entreprises, et il n’est pas tenté ici de les décrire tous en détail. Toutefois, par souci de simplicité, on peut considérer comme règle générale que les équipements de base des procédés hémihydrate et des procédés combinés sont les mêmes, les différences portant uniquement sur les paramètres techniques, les équipements additionnels, l’implantation générale de l’unité et, dans une certaine mesure, les matériaux de construction utilisés.
Procédé hémihydrate — HH
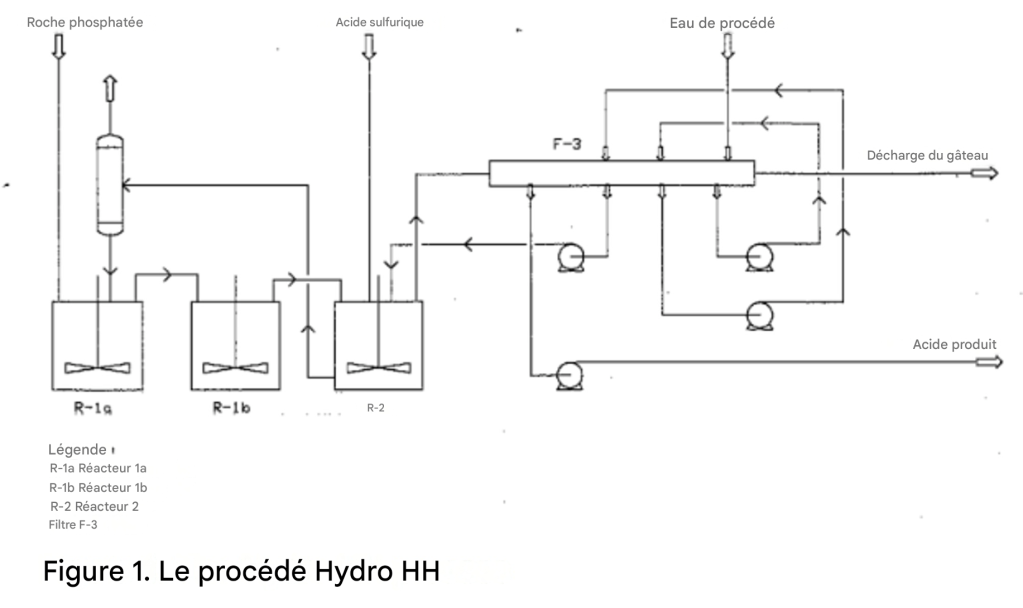
La réaction hémihydrate se déroule dans deux zones distinctes. Il est donc nécessaire de disposer d’au moins deux réacteurs séparés, ou deux compartiments, dans la section réactionnelle. Le rapport volumique préféré est de 2:1. La première zone est souvent divisée en deux compartiments ou réacteurs identiques, 1A et 1B.
La roche phosphatée est introduite dans le réacteur 1A ; l’acide sulfurique et l’acide phosphorique dilué provenant du filtre sont introduits dans le réacteur 2 (Figure 1). La bouillie provenant du réacteur 2 est recyclée à travers un refroidisseur flash vers le réacteur 1A, exposant ainsi la roche phosphatée aux ions sulfate dans des conditions chimiques contrôlées. La bouillie déborde du compartiment 1A vers le compartiment 1B. La chaleur est évacuée par refroidissement sous vide, ou par air si le refroidisseur flash est omis, afin de maintenir la température de la bouillie réactionnelle entre 98 et 100 °C.
L’acide produit, dont la concentration est comprise entre 40 % et 50 % de P₂O₅, selon la qualité et la composition du phosphate, les exigences en aval et le gypse hémihydrate formé, est séparé au moyen d’un filtre horizontal sous vide comportant trois étages de lavage à contre-courant. L’acide produit issu du filtre passe directement vers le stockage. La plupart du temps, il ne nécessite ni clarification ni élimination des solides, et peut être utilisé directement comme acide concentré sans évaporation.
Cependant, certains inconvénients ont eu tendance à limiter la popularité de la voie hémihydrate, parmi lesquels :
Pertes en P₂O₅ et bilan hydrique : le bilan hydrique est plus critique ; par conséquent, la quantité d’eau de lavage pouvant être utilisée est limitée. Les quantités de P₂O₅ soluble et cocristallisé restant dans le gâteau de filtration sont plus importantes, en raison de la concentration plus élevée en P₂O₅ de la bouillie filtrée.
Entartrage : l’hémihydrate n’est pas une forme stable du sulfate de calcium, et il a tendance à se reconvertir en gypse avant même que l’acide ne soit filtré. Pendant le lavage, les conditions sont encore plus favorables à la réhydratation, ce qui peut entraîner l’entartrage des tuyauteries et des équipements, ainsi que le colmatage de la toile filtrante.
Corrosion : à température et concentration acide plus élevées dans un système réactionnel hémihydrate, l’usure des équipements est plus rapide, en particulier celle des agitateurs et des pompes à bouillie.
Toutefois, outre la réduction ou l’élimination du besoin de chaleur d’évaporation, le procédé présente certains avantages :
Économie d’investissement : moins d’équipements d’évaporation sont nécessaires, voire aucun.
Acide plus pur : l’acide contient nettement moins de sulfate libre et de solides en suspension que l’acide du procédé dihydrate évaporé de même concentration, ainsi que des teneurs quelque peu plus faibles en aluminium et en fluor.
Exigences réduites en broyage de la roche : dans des conditions réactionnelles plus sévères, la roche phosphatée réagit beaucoup plus rapidement, et une vitesse de réaction satisfaisante peut être obtenue à partir d’une roche beaucoup plus grossière.
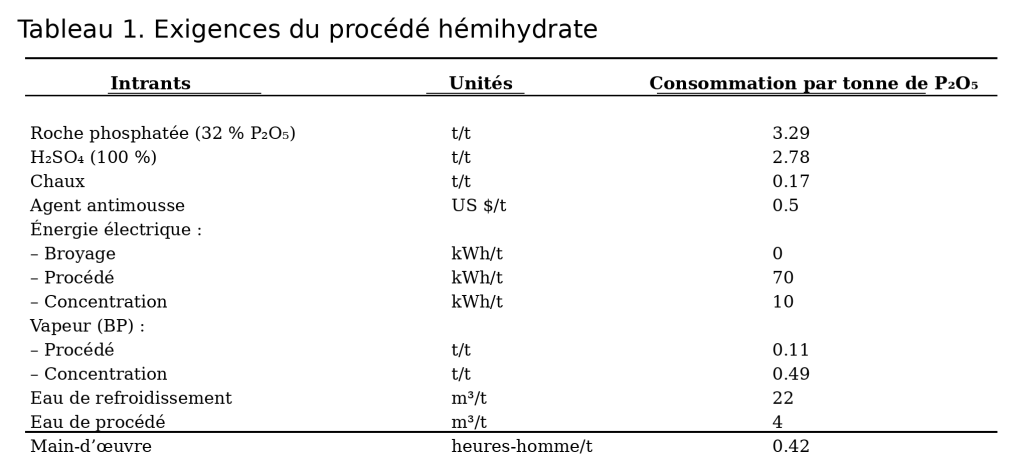
Les consommations moyennes des procédés hémihydrate sont indiquées dans le tableau 1.
BP: basse pression
Procédés hémidihydrate
Les procédés hémihydrate-dihydrate sans filtration intermédiaire, appelés procédés hémihydrate avec recristallisation — HRC —, sont largement utilisés au Maroc, dans les pays d’Asie de l’Est et en Océanie. Les schémas d’implantation de ces unités ressemblent à ceux des procédés dihydrate à réacteurs multiples. Une exception réside dans le fait que les réacteurs d’attaque et de digestion fonctionnent dans des conditions hémihydrate, tandis que les réacteurs suivants fonctionnent dans des conditions favorisant la réhydratation de l’hémihydrate en gypse. Cette transformation est favorisée par des cristaux germes de dihydrate recyclés dans la bouillie provenant de l’alimentation du filtre.
L’acide produit n’est pas plus concentré que celui obtenu par le procédé dihydrate, en raison de la nécessité de cristalliser des cristaux facilement filtrables en présence de l’acide produit. En revanche, le gypse est beaucoup plus pur et présente donc un intérêt plus important, par exemple pour la fabrication du plâtre ou comme retardateur de prise du ciment. Cet avantage est important pour les pays où le gypse naturel est rare, totalement ou partiellement importé, et coûteux.
Le procédé permet également une récupération très élevée du P₂O₅ à partir de la roche, puisque les pertes dans le gypse sont très faibles. La valeur de l’augmentation de 2 à 3 % de la récupération du P₂O₅ dépend du coût de la roche. Les inconvénients sont le coût plus élevé et la complexité accrue du procédé. Par ailleurs, certaines roches phosphatées contiennent des impuretés, telles que les lanthanides, qui stabilisent l’hémihydrate, empêchant ainsi la recristallisation en gypse à une vitesse acceptable.
Procédé hémihydrate-dihydrate avec filtration intermédiaire — HDH
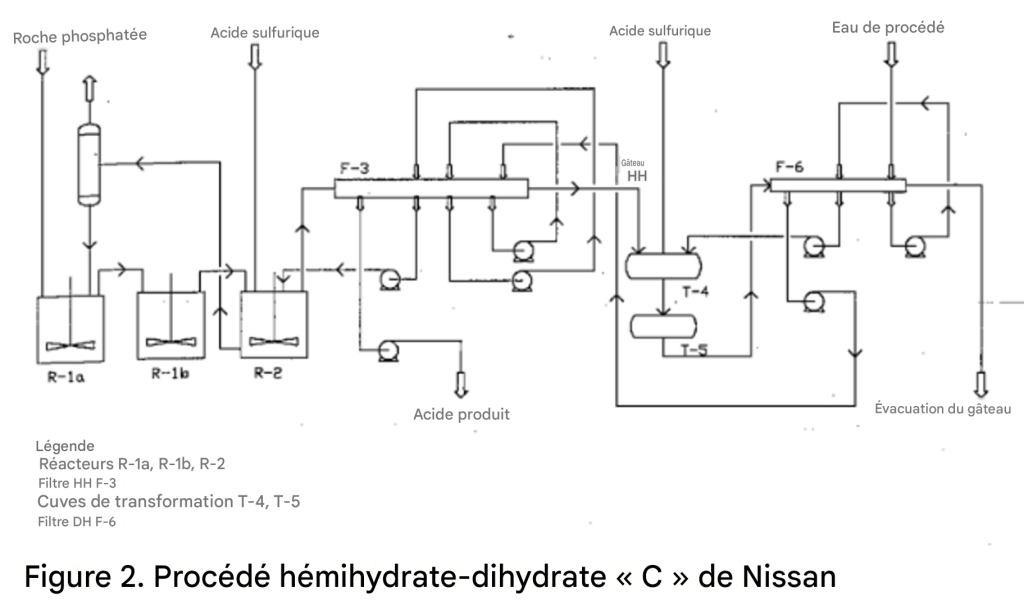
La première étape du procédé est presque identique au procédé HH déjà décrit. Dans l’étape de transformation, le gâteau d’hémihydrate est déchargé du filtre dans une cuve agitée (Figure 2). Les conditions opératoires sont contrôlées afin d’assurer la transformation complète du sulfate de calcium hémihydrate en dihydrate et de laisser suffisamment de temps pour la croissance des cristaux de dihydrate.
La vitesse de transformation est augmentée par l’addition d’une petite alimentation en acide sulfurique. La quasi-totalité du P₂O₅ réticulaire coprécipité avec l’hémihydrate est libérée dans la phase liquide. Le gypse dihydrate est ensuite filtré et le gâteau est lavé avec de l’eau de procédé ou de l’eau de bassin. Le filtrat et le P₂O₅ libéré sont renvoyés vers l’étape de réaction hémihydrate comme dernier lavage sur le filtre hémihydrate.
L’étape supplémentaire de filtration augmente le coût et la complexité de l’unité, mais cet inconvénient peut être compensé par la diminution ou l’élimination de l’étape de concentration, en raison de la forte concentration en P₂O₅, d’environ 45 %, dans l’acide phosphorique produit. De plus, le gypse dihydrate obtenu est beaucoup plus pur que celui issu des procédés HH ou DH, et similaire au gypse HRC ; il peut donc être utilisé dans la production de matériaux de construction.
Les exigences moyennes du procédé hémidihydrate sont indiquées dans le tableau 2.
Procédé dihydrate-hémihydrate — DHH
Bien que les sections d’attaque et de digestion fonctionnent sous conditions dihydrate, il n’est pas souhaitable d’obtenir un degré très élevé de récupération du P₂O₅ lors de la séparation de l’acide du dihydrate, car l’étape ultérieure de déshydratation nécessite environ 20 à 40 % de P₂O₅ et 10 à 20 % de H₂SO₄. Un filtre ou une centrifugeuse peut être utilisé pour produire une bouillie épaissie de gypse dans de l’acide phosphorique à la concentration correcte, et il n’est pas d’une importance critique de produire des cristaux hautement filtrables lors de la première étape. Ainsi, il est possible de produire un acide atteignant environ 35 % de P₂O₅, contre un maximum d’environ 30 % pour le procédé dihydrate ordinaire. La transformation du dihydrate en hémihydrate n’est pas gênée par les impuretés présentes dans la kola-apatite et dans d’autres roches ignées, pour lesquelles ce procédé est très adapté.
Le procédé dihydrate-hémihydrate présente à peu près les mêmes avantages et inconvénients que le procédé hémihydrate-dihydrate, sauf que le sous-produit hémihydrate peut être plus utile que le gypse dans certains cas. De plus, l’hémihydrate présente un comportement d’auto-séchage. Le sulfate de calcium hémihydrate, après filtration, absorbe l’humidité du gâteau pour recristalliser en dihydrate. En revanche, la concentration de l’acide produit est légèrement plus faible.
Un procédé sans filtration intermédiaire, appelé attaque dihydrate – filtration hémihydrate — D-A-HF —, a été développé récemment. Les principaux avantages de ce procédé sont la réduction du coût de l’unité par rapport au procédé DHH à deux étapes de filtration, sa concentration plus élevée en P₂O₅ dans l’acide et sa meilleure récupération du P₂O₅ par comparaison avec le procédé DH classique à une seule étape. En revanche, l’acide produit contient davantage de sulfate, et une étape supplémentaire de désulfatation peut être nécessaire selon l’application en aval.
Ingénierie / construction de l’unité
Matériaux de construction: Le choix des matériaux de construction est très important, car il influence le degré de maintenance et les temps d’arrêt d’une unité de production d’acide phosphorique.
Les vitesses de corrosion dans une unité sont variables et dépendent largement de la teneur en chlorures et en fluorures libres de l’acide.
Les matériaux de construction typiques pour les différents équipements sont les suivants :
Réacteurs : béton, avec agrégats résistants aux acides tels que le sable siliceux et le gravier ; acier doux revêtu de caoutchouc — RLMS, rubber lined mild steel — protégé par des briques de carbone.
Cuves : RLMS, polypropylène, FRP — polypropylène renforcé de fibres.
Tuyauteries : polypropylène, FRP, acier carbone revêtu de caoutchouc.
Agitateurs : Alloy 20, 904L, HV9, Sanicro 28, Ferralium 255, 317L, Uranus B6 et Uranus 52N+ — Super Duplex.
Pompes : Alloy 20 et 904L, Alloy 28, HV9, Sanicro 28, polyéthylène haute densité, Ferrahum 255, 317L, Hastelloy C et Uranus 52N+ — Super Duplex.
Échangeurs de chaleur : graphite imprégné renforcé par fibre de carbone, tel que Diabon — Sigri —, Bigilor, Graphilor BS — Le Carbone-Lorraine.
Les parties les plus critiques et les plus vulnérables d’une unité d’acide phosphorique sont les agitateurs, les pompes et les filtres. Les conditions corrosives sont les plus sévères pour les agitateurs ; toutefois, ceux-ci peuvent être remplacés avec une relative facilité et à faible coût. Les filtres, en revanche, sont volumineux, coûteux et difficiles à remplacer ou à réparer. Les conditions corrosives sont un peu moins sévères pour les filtres que pour les agitateurs, car la température moyenne y est plus basse et l’érosion constitue un facteur moins important.
Pendant de nombreuses années, l’acier inoxydable 316L a été un matériau de construction courant pour les agitateurs, les filtres et les autres équipements en contact direct avec l’acide phosphorique humide ou la bouillie réactionnelle. Il est maintenant remplacé par le 904L pour l’acide et par l’Uranus 52N+ — Super Duplex — pour les bouillies abrasives.
192, 1980, The Oxy Hemihydrate Phosphoric Acid Process, M. B. Caesar, H. C. Smith, L. E. Mercando.
209, 1982, Recent Experiences in Phosphoric Acid Production by Hemihydrate Routes, M. L. Parker, C. McDonald.
248, 1986, Relative Merits of Different Filters for Hemihydrate Filtration, M. L. Parker, J. A. Hallsworth.
Liens vers des sources externes
Becker, P. 1989. Phosphates and Phosphoric Acid: Raw Materials, Technology, and Economics of the Wet Process. Marcel Dekker, Inc., New York, NY, U.S.A.
Havelange, S. et al. 2022. Phosphoric Acid and Phosphates dans Ullmann’s Encyclopaedia of Industrial Chemistry.
Slack, A. V. 1968. Phosphoric Acid — Part I and II. Marcel Dekker, Inc., New York, NY, U.S.A.