Gerald Steiner, Bernhard Geissler, and Nils Haneklaus
Introduction
Les États-Unis sont le plus grand consommateur mondial d’uranium. En 2017, la très grande majorité, soit 93 %, de cet uranium était importée. Une enquête récente menée par l’administration actuelle a conclu que les importations étrangères d’uranium et l’approvisionnement en produits connexes — essentiels pour l’arsenal nucléaire américain, la marine hauturière et les centrales électriques — ne constituaient pas une menace pour la sécurité nationale. Toutefois, le déclin de l’exploitation minière nationale de l’uranium est considéré comme une préoccupation importante.
Les exploitants miniers d’uranium aux États-Unis ont lancé cette enquête dans l’espoir d’obtenir des quotas sur les importations étrangères d’uranium, ce qui leur permettrait de mieux concurrencer les entreprises étrangères, souvent publiques et fortement subventionnées. Des quotas, s’ils étaient mis en place, pourraient en effet relancer l’exploitation minière de l’uranium aux États-Unis. Ils pourraient également redonner un intérêt à la récupération de l’uranium à partir de ressources non conventionnelles, notamment les phosphates, qui, à leur apogée, ont contribué à près de 20 % des besoins du pays en uranium dans les années 1980. Cette voie pourrait redevenir rentable et être mise en œuvre beaucoup plus rapidement que l’ouverture de nouvelles mines d’uranium, lesquelles finiraient nécessairement par devoir être implantées quelque part, avec les oppositions locales que cela implique.
Les phosphates — et plus précisément ici les roches phosphatées sédimentaires — peuvent contenir des quantités considérables d’uranium associé, aussi bien en termes de concentrations que de quantités globales. La roche phosphatée est le quatrième matériau le plus extrait au monde et elle est principalement utilisée, à plus de 90 % à l’échelle mondiale, pour la production d’engrais minéraux.
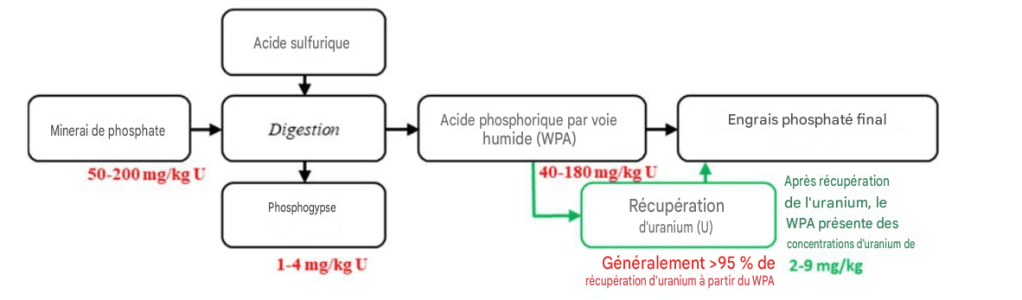
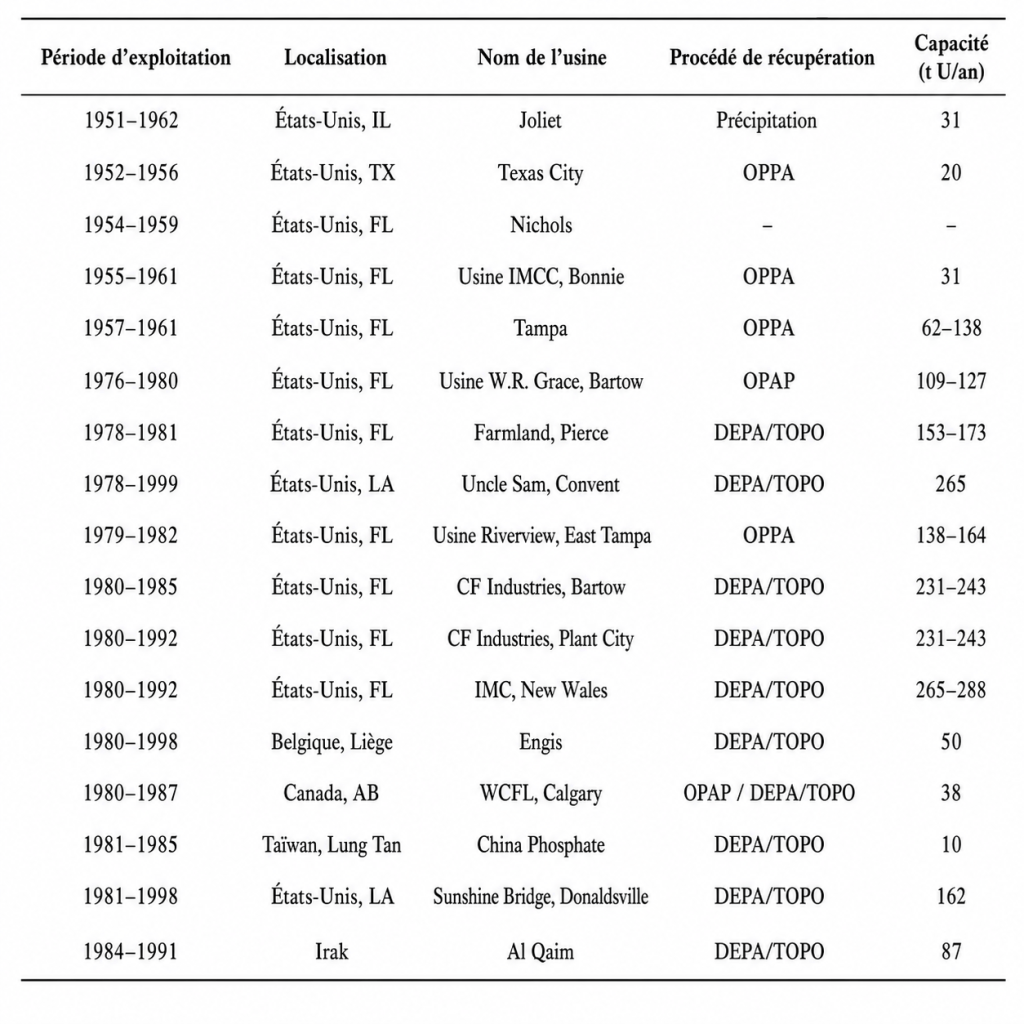
Les techniques permettant de récupérer l’uranium à partir de l’acide phosphorique, produit intermédiaire liquide de la fabrication des engrais phosphatés, sont bien connues et ont été utilisées aux États-Unis, ainsi que, dans une moindre mesure, ailleurs, à l’échelle industrielle jusqu’à la fin des années 1990, lorsque les prix de l’uranium se sont effondrés, rendant la récupération non rentable pour les producteurs d’engrais.
Quantités d’uranium dans les phosphates
La sensibilisation accrue aux enjeux environnementaux, la sécurité énergétique nationale et la possible hausse des prix de l’uranium ont suscité un regain d’intérêt mondial pour cette technologie. Gabriel et al. ainsi qu’Ulrich et al. ont estimé que les producteurs d’engrais phosphatés pourraient fournir un peu plus de 15 % des besoins mondiaux en uranium en temps de paix. Des études similaires ont été réalisées pour l’Argentine, où l’uranium récupéré comme coproduit de la production d’engrais phosphatés pourrait couvrir 8 à 9 % des besoins en uranium ; pour l’Union européenne, environ 2 % des besoins en uranium ; et pour les États-Unis, environ 10 % des besoins en uranium.
Dans ces études, les quantités d’uranium récupérables à partir des phosphates dépasseraient souvent la production actuelle issue des mines d’uranium conventionnelles nationales. Kim et al. ont par exemple estimé que 5,5 millions de livres de U₃O₈, soit davantage que la production nationale américaine de 2014, qui était de 4,9 millions de livres de U₃O₈, auraient pu être fournies par l’industrie américaine des phosphates.
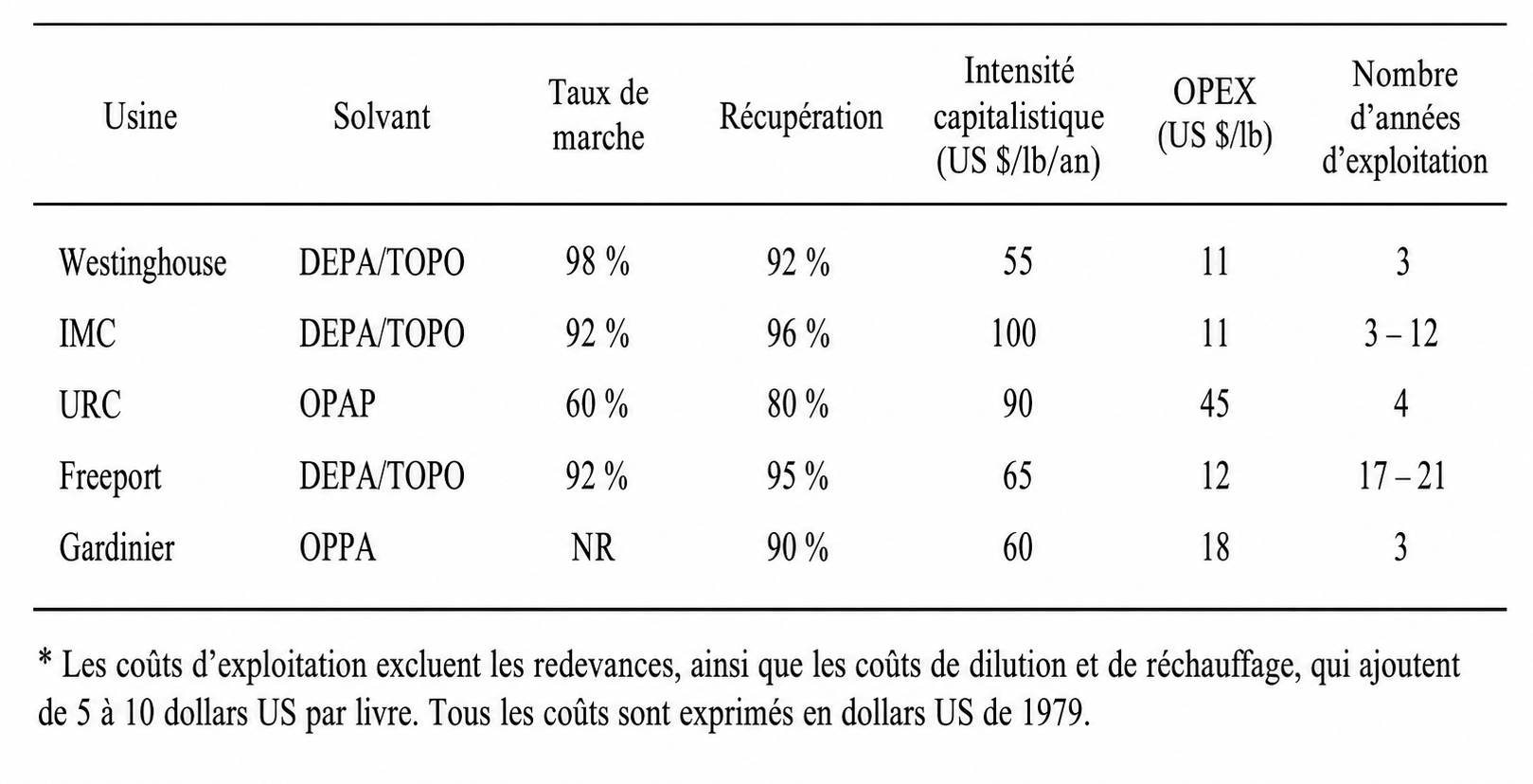
La figure 1 compare la production annuelle d’uranium aux importations annuelles d’uranium aux États-Unis. Elle indique également la récupération historique de l’uranium à partir des phosphates ainsi que le potentiel de récupération de l’uranium à partir des phosphates aux États-Unis. Avec la diminution de l’exploitation minière de l’uranium en 1990, les quantités d’uranium qui auraient pu être récupérées à partir des phosphates étaient supérieures aux quantités effectivement extraites des mines.
Dans la figure 1, sont pris en compte l’uranium provenant de toutes les mines de phosphate des États-Unis ainsi que l’uranium contenu dans les importations de roches phosphatées. Bien que sa concentration soit relativement élevée en Floride, environ 160 mg/kg — à comparer à la mine de Rössing en Namibie, l’une des plus anciennes exploitations commerciales d’uranium en activité et la cinquième plus grande mine d’uranium commerciale, avec des concentrations moyennes d’uranium de 200 à 300 mg/kg et une teneur moyenne de 0,003 mg/kg d’uranium dans l’eau de mer — cette teneur justifierait une récupération. Toutefois, la concentration peut être moins élevée dans d’autres régions, par exemple 107 mg/kg dans l’Idaho et 65 mg/kg en Caroline du Nord. Dans ces conditions, une récupération rentable de l’uranium serait plus difficile.
Dans ce contexte, il convient de noter que la majorité de l’uranium radiotoxique, soit 70 à 80 %, qui n’est pas récupéré est — et continuera d’être — dispersée avec les engrais sur les sols agricoles. Le reste se retrouve dans le flux de déchets de phosphogypse, c’est-à-dire une boue composée de sulfate de calcium et d’eau, présentant de faibles niveaux de radioactivité et donc des possibilités d’utilisation limitées.
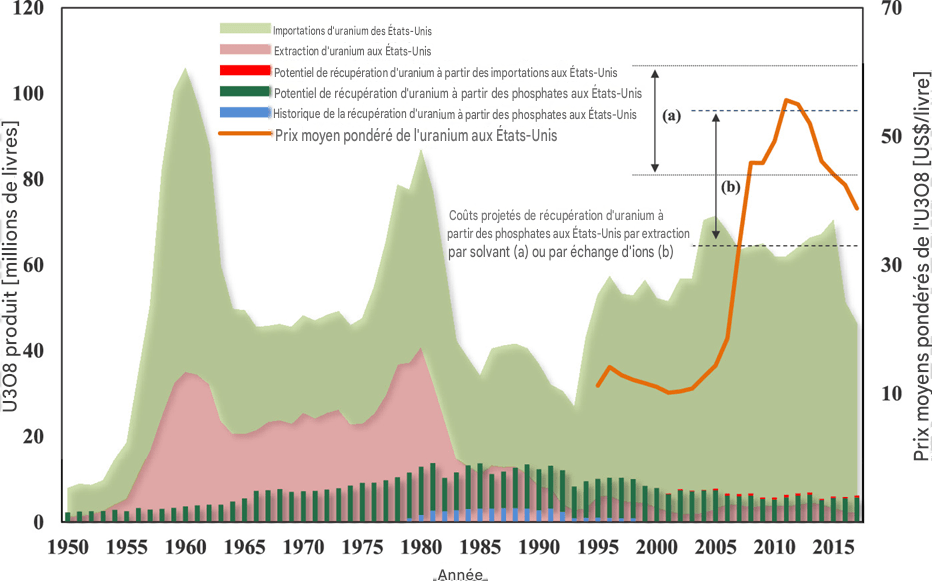
Figure 1. Importations américaines d’uranium, production nationale d’uranium, récupération potentielle et historique de l’uranium à partir des phosphates, prix moyens pondérés de l’uranium, ainsi que coûts minimaux et maximaux projetés pour la récupération de l’uranium par extraction par solvant (a) et par échange d’ions (b) aux États-Unis.
Source : EIA (8) et Beltrami et al. (1).
Économie de la récupération
Malgré des prix de l’uranium relativement faibles, les propriétaires et exploitants de réacteurs électronucléaires civils aux États-Unis ont acheté, en 2018, un total de 40 millions de livres d’équivalent U₃O₈ auprès de fournisseurs américains et étrangers, à un prix moyen pondéré de 38,81 dollars américains par livre de U₃O₈. Près de 42 % de cet uranium provenait du Canada et d’Australie ; environ 40 % provenait du Kazakhstan, de Russie et d’Ouzbékistan ; et près de 10 % provenait des États-Unis. L’uranium d’origine américaine a été livré à un prix moyen pondéré de 45,26 dollars américains par livre de U₃O₈.
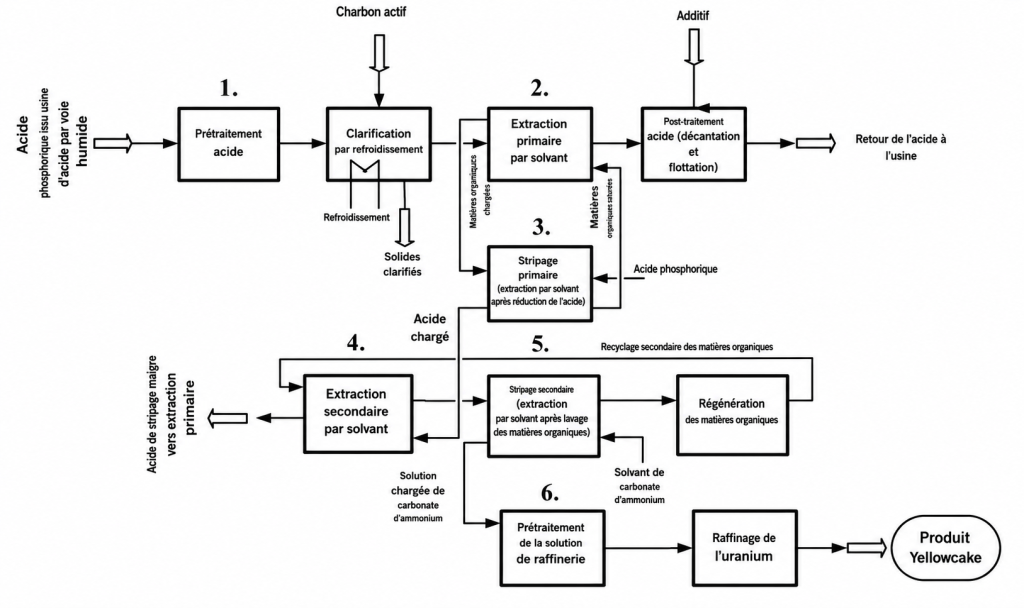
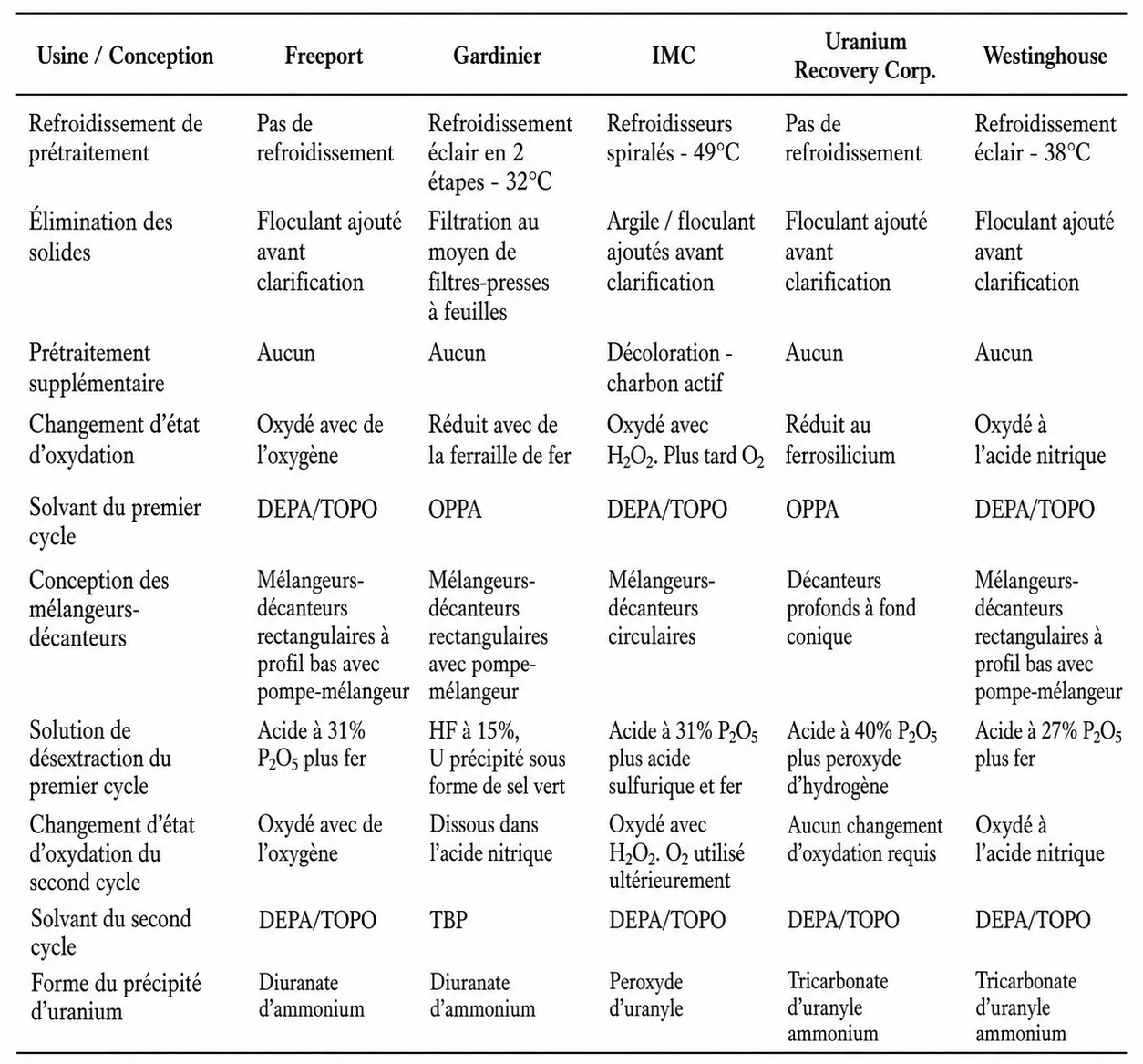
Dans ce contexte, les coûts de récupération de l’uranium à partir de l’acide phosphorique, au moyen d’une technologie d’extraction par solvants industriellement éprouvée, sont estimés aujourd’hui aux États-Unis entre 44 et 61 dollars américains par livre de U₃O₈. Ces coûts se situent donc à la limite de la rentabilité économique.
La récupération de l’uranium au moyen de procédés fondés sur l’échange d’ions n’est pas encore une technologie commerciale éprouvée pour la récupération de l’uranium, mais elle est actuellement testée à l’échelle pilote aux États-Unis par PhosEnergy, une société australienne. Elle pourrait réduire davantage les coûts, jusqu’à environ 33 à 54 dollars américains par livre de U₃O₈.
Des économies encore plus importantes pourraient être réalisées si l’uranium radiotoxique était lixivié directement, avant l’étape d’attaque/digestion, à partir de la roche phosphatée enrichie. Cela permettrait de rendre non seulement l’engrais final, mais aussi le sous-produit phosphogypse, pratiquement exempts d’uranium.
Bien que le gypse soit un matériau de construction largement utilisé, le phosphogypse est généralement stocké indéfiniment en raison de sa faible radioactivité, qui résulte de la présence naturelle d’uranium et, dans une moindre mesure, de thorium dans les roches phosphatées traitées. Quelque 100 à 300 millions de tonnes de phosphogypse sont produites chaque année dans le monde. Rendre ce matériau disponible pour des utilisations non contestables pourrait donc permettre à l’industrie des engrais d’économiser des centaines de millions de dollars américains en coûts annuels de stockage qui s’accumulent autrement.
Par ailleurs, au regard des coûts directs précédemment mentionnés pour la récupération de l’uranium à partir des phosphates, qui peuvent être estimés aujourd’hui, il convient de souligner que nous pourrions être confrontés à des coûts indirects croissants liés à la purification des eaux souterraines contaminées par l’uranium issu des engrais si les pratiques actuelles restent inchangées.
Quo vadis, récupération de l’uranium ?
La récupération de l’uranium à partir des phosphates est déjà une bonne idée du point de vue de la conservation des ressources et des Objectifs de développement durable, ODD, des Nations unies. Le cadre établi par les décideurs politiques américains déterminera si l’uranium sera ou non à nouveau récupéré au cours de la production d’engrais.
Dans ce contexte, des quotas sur les importations étrangères d’uranium aux États-Unis pourraient, de manière consciente ou non, fournir un cadre incitatif à la récupération de l’uranium pendant la production américaine d’engrais, et ainsi rendre à nouveau attractive la récupération de l’uranium à partir des phosphates.
Informations sur les auteurs
Auteur correspondant
Nils Haneklaus — Danube University Krems, Krems, Autriche, et RWTH Aachen University, Aix-la-Chapelle, Allemagne ;
ORCID : orcid.org/0000-0002-673-0376 ;
Email : nils.haneklaus@rwth-aachen.de
Autres auteurs
Gerald Steiner — Danube University Krems, Krems, Autriche
Bernhard Geissler — Danube University Krems, Krems, Autriche
Les informations complètes de contact sont disponibles à l’adresse suivante :
https://pubs.acs.org/10.1021/acs.est.9b07859
Notes
Les auteurs déclarent ne pas avoir d’intérêts financiers concurrents.
Références
(1) Beltrami, D.; Cote, G.; Mokhtari, H.; Courtaud, B.; Moyer, B. A.; Chagnes, A. Recovery of Uranium from Wet Process Phosphoric Acid by Solvent Extraction. Chem. Rev. 2014, 114, 12002–12023.
(2) Singh, D. K.; Mondal, S.; Chakravartty, J. K. Recovery of Uranium From Phosphoric Acid: A Review. Solvent Extr. Ion Exch. 2016, 34, 201–25.
(3) Gabriel, S.; Baschwitz, A.; Mathonnière, G.; Fizaine, F.; Eleouet, T. Building future nuclear power fleets: The available uranium resources constraint. Resour. Policy 2013, 38, 458–469.
(4) Ulrich, E.; Schnug, H.-M.; Prasser, E. Frossard Uranium endowments in phosphate rock. Sci. Total Environ. 2014, 478, 226–234.
(5) López, L.; Castro, L. N.; Scasso, R. A.; Grancea, L.; Tulsidas, H.; Haneklaus, N.; Nacional, C.; Atamica, D. E.; Libertador, A.; Aires, C. B. Uranium supply potential from phosphate rocks for Argentina’s nuclear power fleet. Resour. Policy 2019, 62, 397–404.
(6) Tulsidas, H.; Gabriel, S.; Kiegiel, K.; Haneklaus, N. Uranium resources in EU phosphate rock imports. Resour. Policy 2019, 61, 151–156.
(7) Kim, H.; Eggert, R. G.; Carlsen, B. W.; Dixon, B. W. Potential uranium supply from phosphoric acid: A U.S. analysis comparing solvent extraction and ion exchange recovery. Resour. Policy 2016, 49, 222–231.
(8) EIA. Uranium Marketing Annual Report, 2019; https://www.eia.gov/uranium/marketing/.
(9) Hore-Lacy, I. Production of Byproduct Uranium and Uranium from Unconventional Resources; Elsevier Ltd, 2016; http://dx.doi.org/10.1016/B978-0-08-100307-7.00009-0.
(10) Al Khaledi, N.; Taha, M.; Hussein, A.; Hussein, E.; El Yahyaoui, A.; Haneklaus, N. Direct leaching of rare earth elements and uranium from phosphate rocks. IOP Conf. Ser.: Mater. Sci. Eng. 2019, 479, 012065.